

Regulation of mitochondrial transport in neurons
- PMID: 25612908
- PMCID: PMC4433773
- DOI: 10.1016/j.yexcr.2015.01.004
Regulation of mitochondrial transport in neurons
Abstract
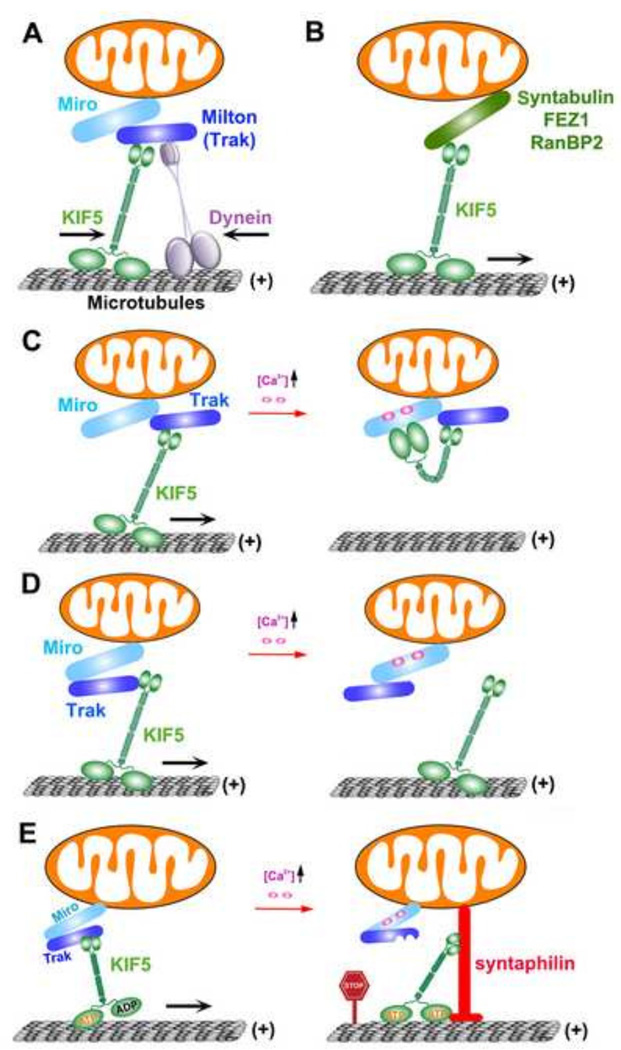
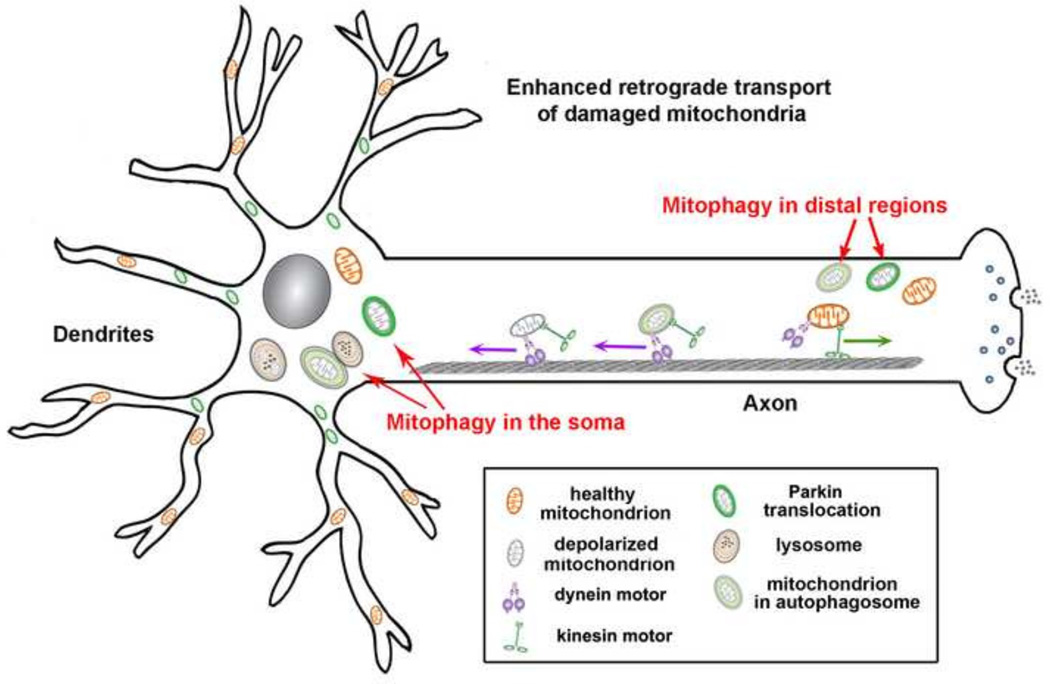
Mitochondria are cellular power plants that supply ATP to power various biological activities essential for neuronal growth, survival, and function. Due to unique morphological features, neurons face exceptional challenges to maintain ATP and Ca(2+) homeostasis. Neurons require specialized mechanisms distributing mitochondria to distal areas where energy and Ca(2+) buffering are in high demand, such as synapses and axonal branches. These distal compartments also undergo development- and activity-dependent remodeling, thereby altering mitochondrial trafficking and distribution. Mitochondria move bi-directionally, pause briefly, and move again, frequently changing direction. In mature neurons, only one-third of axonal mitochondria are motile. Stationary mitochondria serve as local energy sources and buffer intracellular Ca(2+). The balance between motile and stationary mitochondria responds quickly to changes in axonal and synaptic physiology. Furthermore, neurons are postmitotic cells surviving for the lifetime of the organism; thus, mitochondria need to be removed when they become aged or dysfunction. Mitochondria also alter their motility under stress conditions or when their integrity is impaired. Therefore, regulation of mitochondrial transport is essential to meet altered metabolic requirements and to remove aged and damaged mitochondria or replenish healthy ones to distal terminals. Defects in mitochondrial transport and altered distribution are implicated in the pathogenesis of several major neurological disorders. Thus, research into the mechanisms regulating mitochondrial motility is an important emerging frontier in neurobiology. This short review provides an updated overview on motor-adaptor machineries that drive and regulate mitochondrial transport and docking receptors that anchor axonal mitochondria in response to the changes in synaptic activity, metabolic requirement, and altered mitochondrial integrity. The review focuses on microtubule (MT)-based mitochondrial trafficking and anchoring. Additional insight from different perspectives can be found in other in-depth reviews.
Keywords: Dynein motors; Kinesin motors; Mitochondrial docking; Mitochondrial transport; Mitophagy; Motile mitochondria; Stationary mitochondria; Synaptic activity; Syntaphilin.
Published by Elsevier Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
