

Cholesterol, inflammation and innate immunity
- PMID: 25614320
- PMCID: PMC4669071
- DOI: 10.1038/nri3793
Cholesterol, inflammation and innate immunity
Abstract
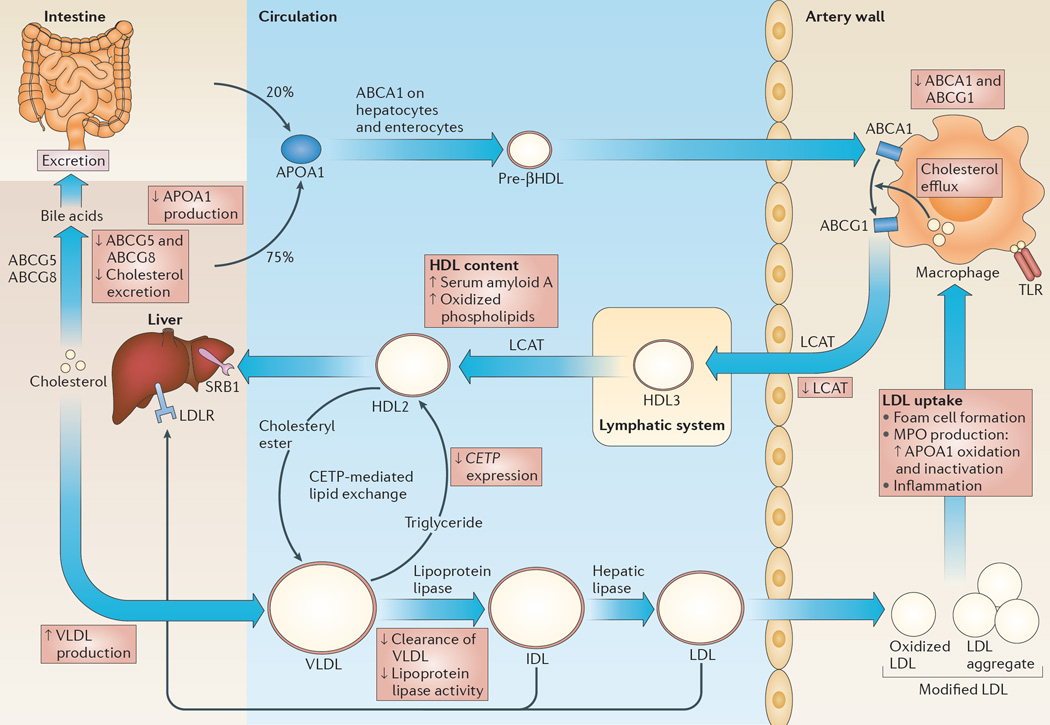
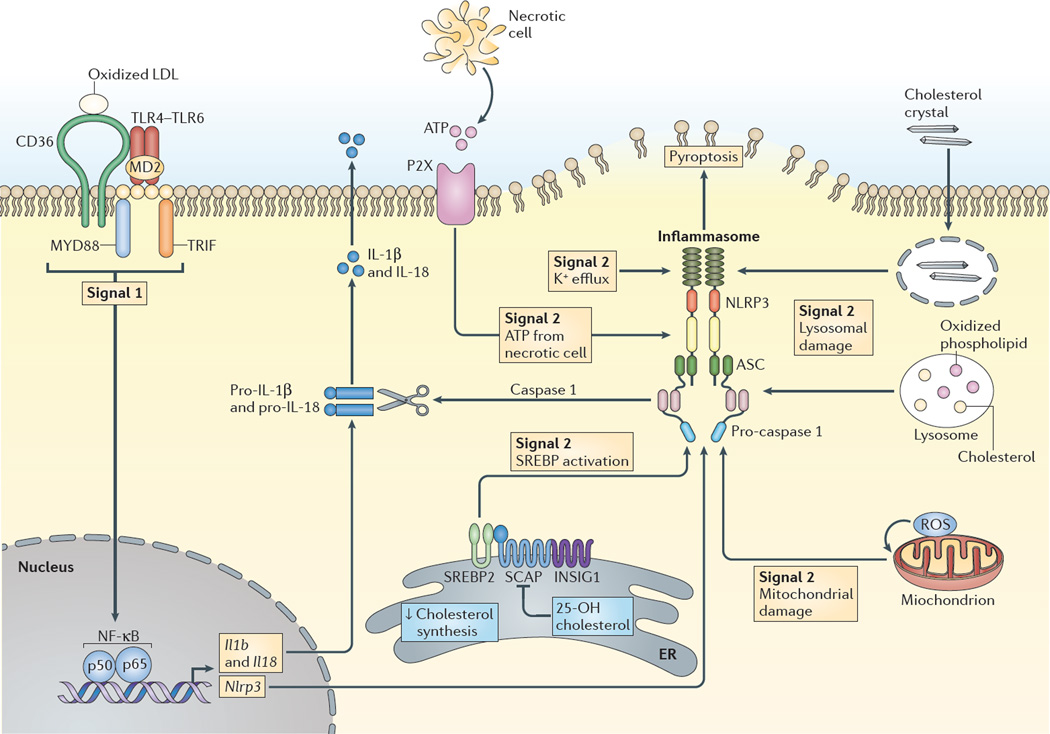
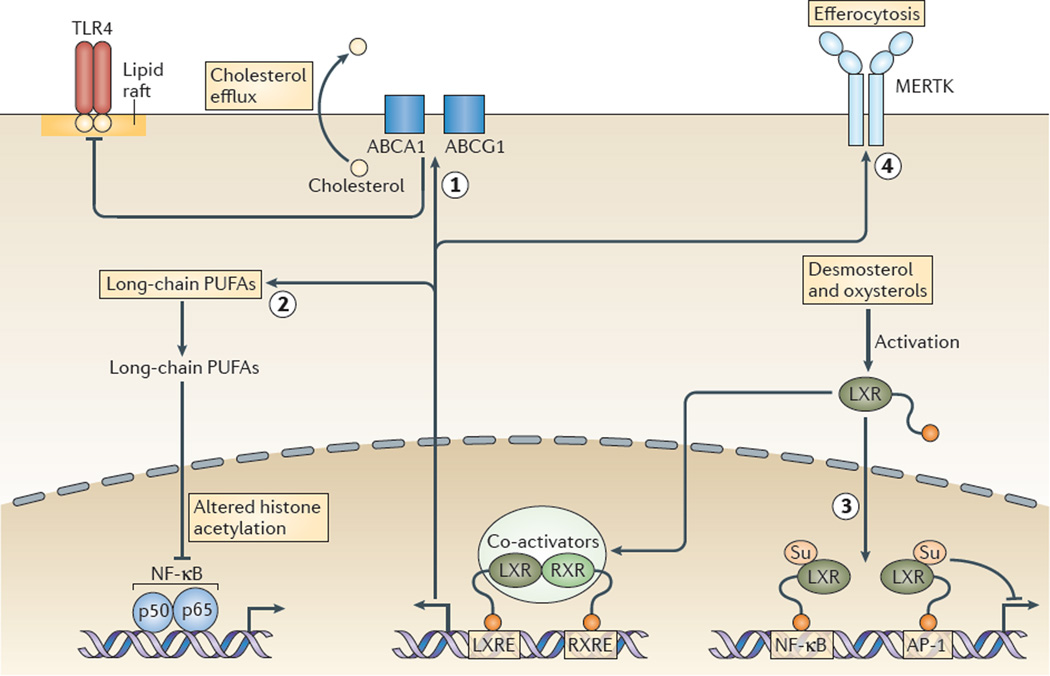
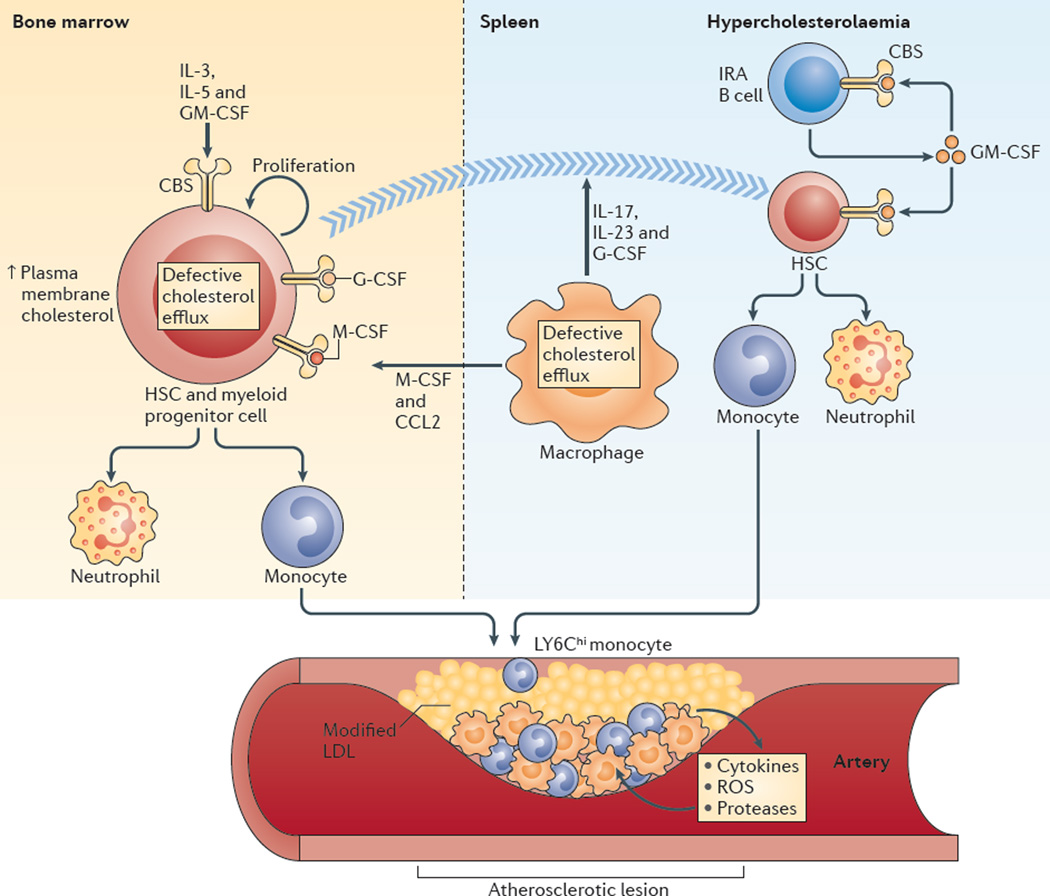
Hypercholesterolaemia leads to cholesterol accumulation in macrophages and other immune cells, which promotes inflammatory responses, including augmentation of Toll-like receptor (TLR) signalling, inflammasome activation, and the production of monocytes and neutrophils in the bone marrow and spleen. On a cellular level, activation of TLR signalling leads to decreased cholesterol efflux, which results in further cholesterol accumulation and the amplification of inflammatory responses. Although cholesterol accumulation through the promotion of inflammatory responses probably has beneficial effects in the response to infections, it worsens diseases that are associated with chronic metabolic inflammation, including atherosclerosis and obesity. Therapeutic interventions such as increased production or infusion of high-density lipoproteins may sever the links between cholesterol accumulation and inflammation, and have beneficial effects in patients with metabolic diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
