

Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions
- PMID: 25615868
- PMCID: PMC4304694
- DOI: 10.1107/S1399004714021683
Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions
Abstract
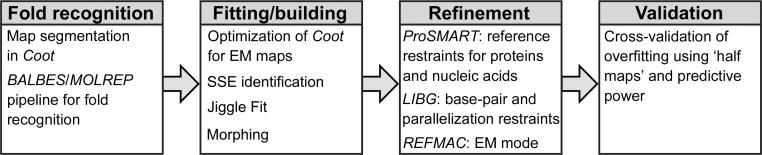
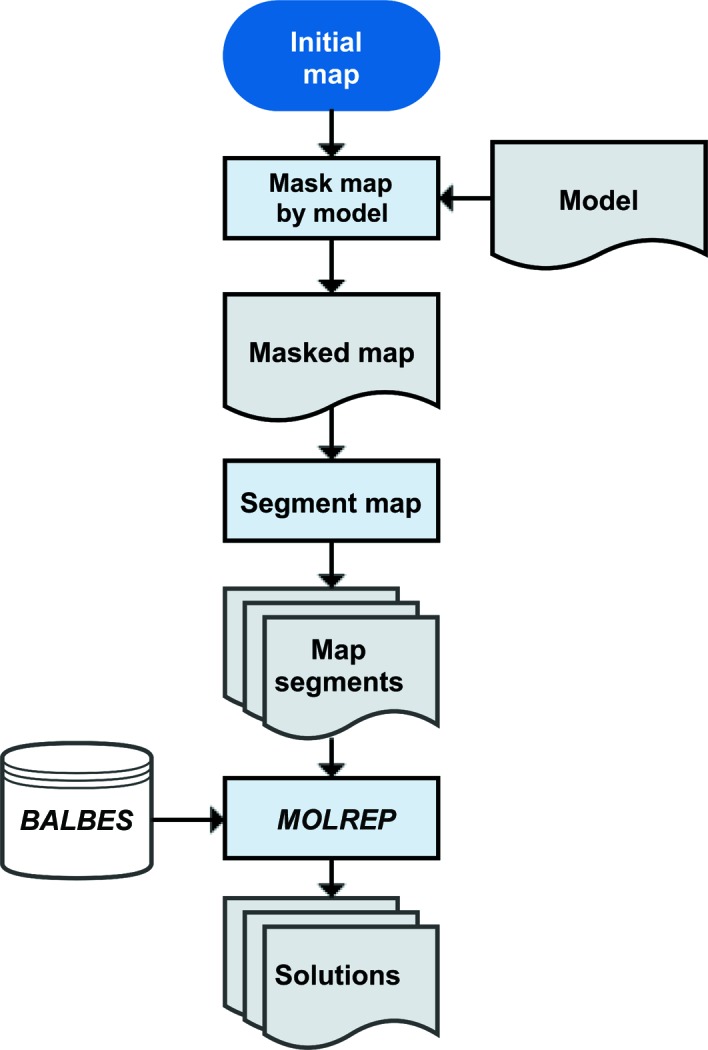
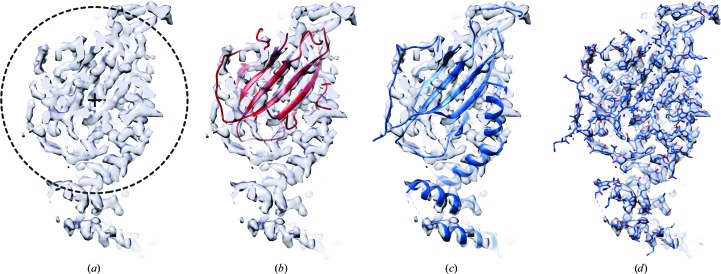
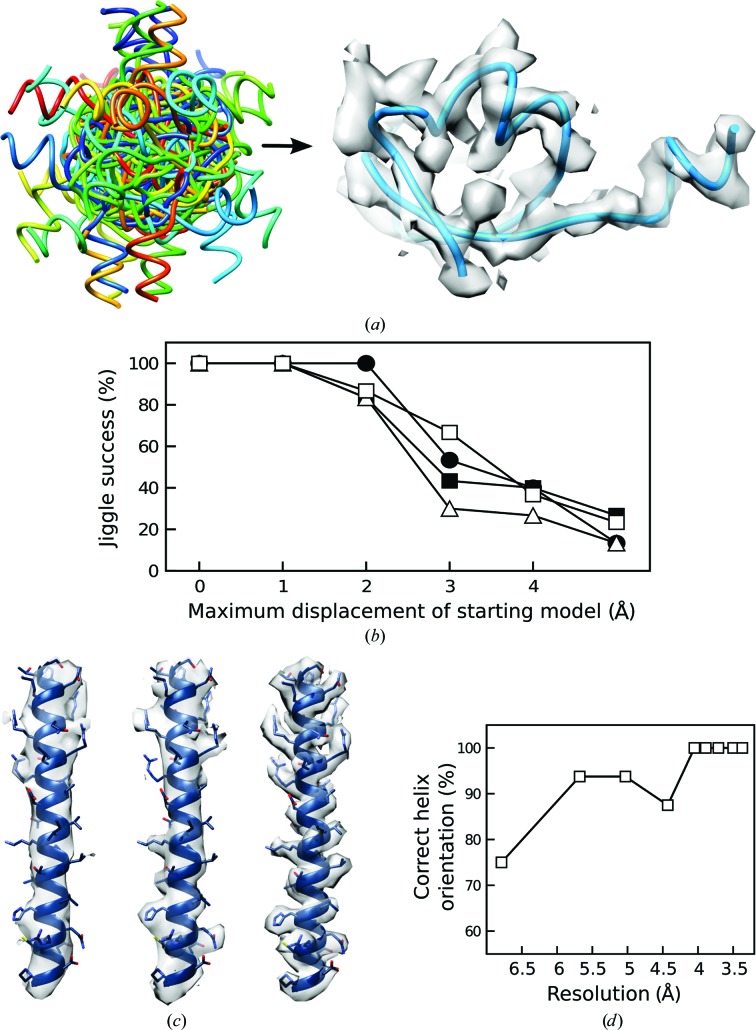
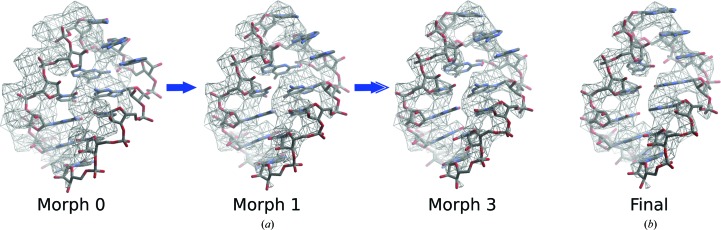
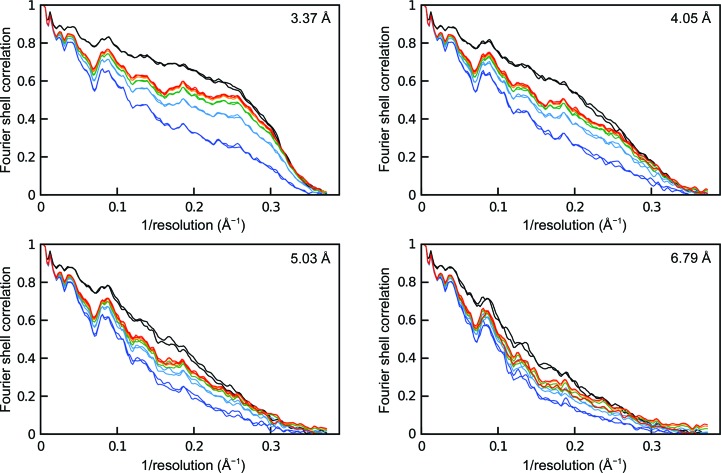
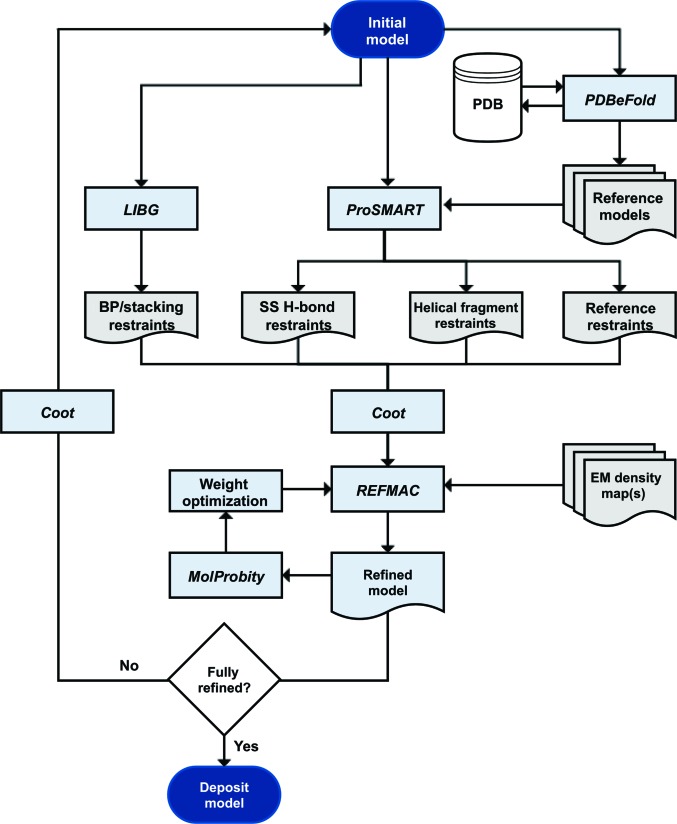
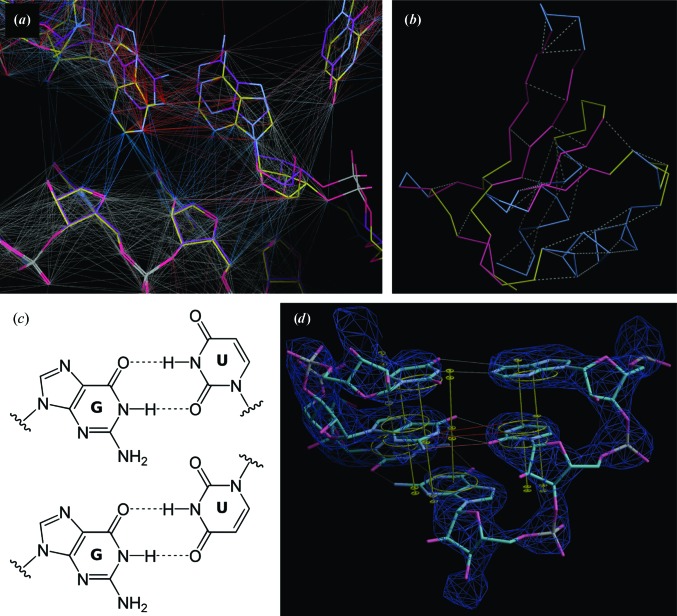
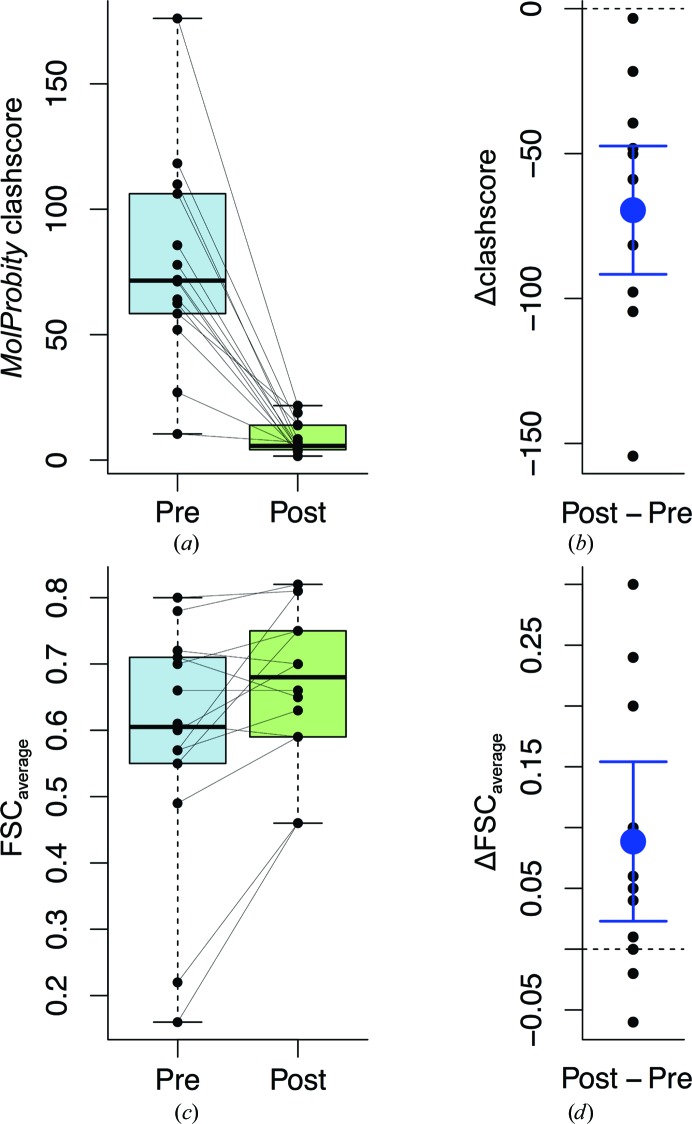
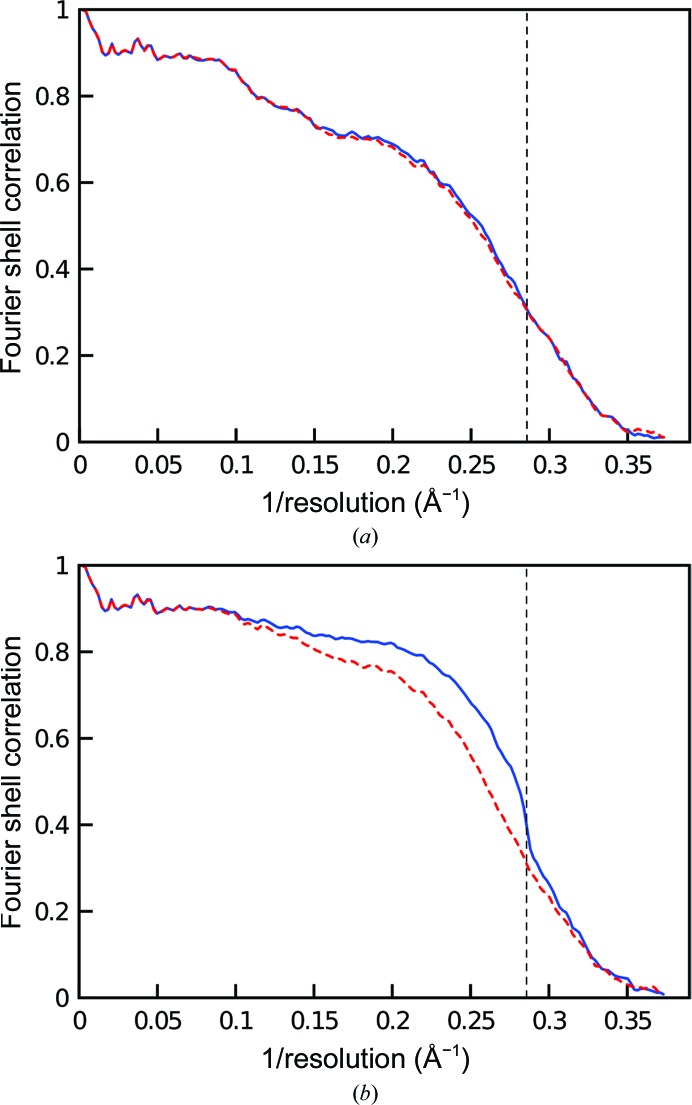
The recent rapid development of single-particle electron cryo-microscopy (cryo-EM) now allows structures to be solved by this method at resolutions close to 3 Å. Here, a number of tools to facilitate the interpretation of EM reconstructions with stereochemically reasonable all-atom models are described. The BALBES database has been repurposed as a tool for identifying protein folds from density maps. Modifications to Coot, including new Jiggle Fit and morphing tools and improved handling of nucleic acids, enhance its functionality for interpreting EM maps. REFMAC has been modified for optimal fitting of atomic models into EM maps. As external structural information can enhance the reliability of the derived atomic models, stabilize refinement and reduce overfitting, ProSMART has been extended to generate interatomic distance restraints from nucleic acid reference structures, and a new tool, LIBG, has been developed to generate nucleic acid base-pair and parallel-plane restraints. Furthermore, restraint generation has been integrated with visualization and editing in Coot, and these restraints have been applied to both real-space refinement in Coot and reciprocal-space refinement in REFMAC.
Keywords: LIBG; electron cryo-microscopy reconstructions; model building; refinement.
Figures
References
-
- Alexandrov, N. & Shindyalov, I. (2003). Bioinformatics, 19, 429–430. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
