

A small molecule PAI-1 functional inhibitor attenuates neointimal hyperplasia and vascular smooth muscle cell survival by promoting PAI-1 cleavage
- PMID: 25617690
- PMCID: PMC4361315
- DOI: 10.1016/j.cellsig.2015.01.009
A small molecule PAI-1 functional inhibitor attenuates neointimal hyperplasia and vascular smooth muscle cell survival by promoting PAI-1 cleavage
Abstract
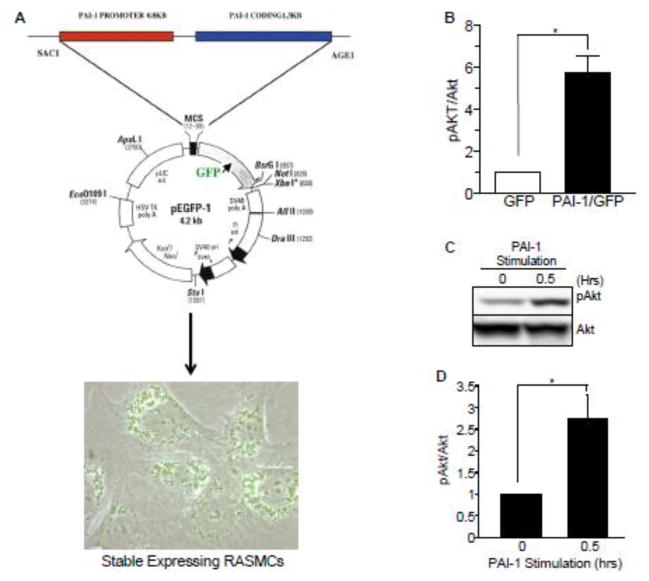
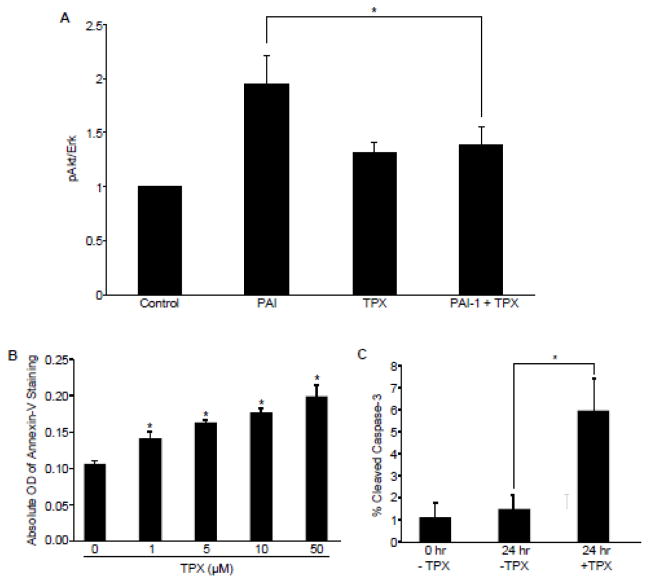
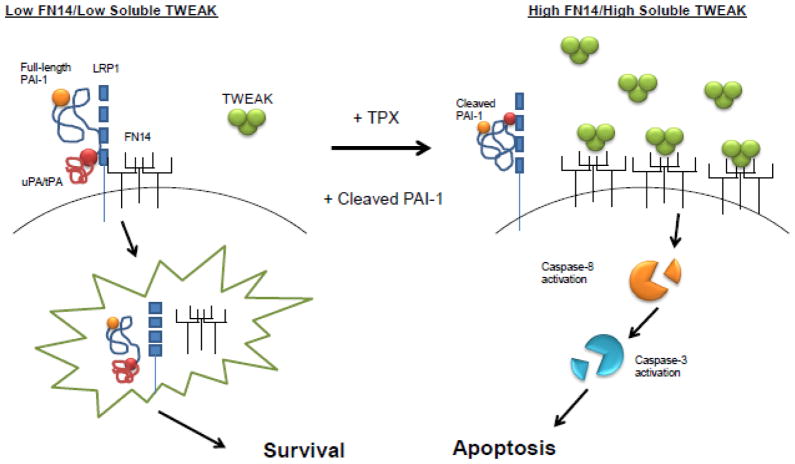
Plasminogen activator inhibitor-1 (PAI-1), the primary inhibitor of urokinase-and tissue-type plasminogen activators (uPA and tPA), is an injury-response gene implicated in the development of tissue fibrosis and cardiovascular disease. PAI-1 mRNA and protein levels were elevated in the balloon catheter-injured carotid and in the vascular smooth muscle cell (VSMC)-enriched neointima of ligated arteries. PAI-1/uPA complex formation and PAI-1 antiproteolytic activity can be inhibited, via proteolytic cleavage, by the small molecule antagonist tiplaxtinin which effectively increased the VSMC apoptotic index in vitro and attenuated carotid artery neointimal formation in vivo. In contrast to the active full-length serine protease inhibitor (SERPIN), elastase-cleaved PAI-1 (similar to tiplaxtinin) also promoted VSMC apoptosis in vitro and similarly reduced neointimal formation in vivo. The mechanism through which cleaved PAI-1 (CL-PAI-1) stimulates apoptosis appears to involve the TNF-α family member TWEAK (TNF-α weak inducer of apoptosis) and it's cognate receptor, fibroblast growth factor (FGF)-inducible 14 (FN14). CL-PAI-1 sensitizes cells to TWEAK-stimulated apoptosis while full-length PAI-1 did not, presumably due to its ability to down-regulate FN14 in a low density lipoprotein receptor-related protein 1 (LRP1)-dependent mechanism. It appears that prolonged exposure of VSMCs to CL-PAI-1 induces apoptosis by augmenting TWEAK/FN14 pro-apoptotic signaling. This work identifies a critical, anti-stenotic, role for a functionally-inactive (at least with regard to its protease inhibitory function) cleaved SERPIN. Therapies that promote the conversion of full-length to cleaved PAI-1 may have translational implications.
Keywords: Apoptosis; Carotid stenosis; PAI-1; SERPIN; Vascular injury.
Copyright © 2015 Elsevier Inc. All rights reserved.
Conflict of interest statement
None declared.
Figures






References
-
- Declerck PJ, De Mol M, Vaughan DE, Collen D. Identification of a conformationally distinct form of plasminogen activator inhibitor-1, acting as a noninhibitory substrate for tissue-type plasminogen activator. J Biol Chem. 1992;267:11693–11696. - PubMed
-
- Urano T, Strandberg L, Johansson LB, Ny T. A substrate-like form of plasminogen-activator-inhibitor type 1. Conversions between different forms by sodium dodecyl sulphate. Eur J Biochem. 1992;209:985–992. - PubMed
-
- Dellas C, Loskutoff DJ. Historical analysis of PAI-1 from its discovery to its potential role in cell motility and disease. Thromb Haemost. 2005;93:631–640. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
