

Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6
- PMID: 25620207
- PMCID: PMC4320262
- DOI: 10.1016/j.ajhg.2014.12.011
Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6
Abstract
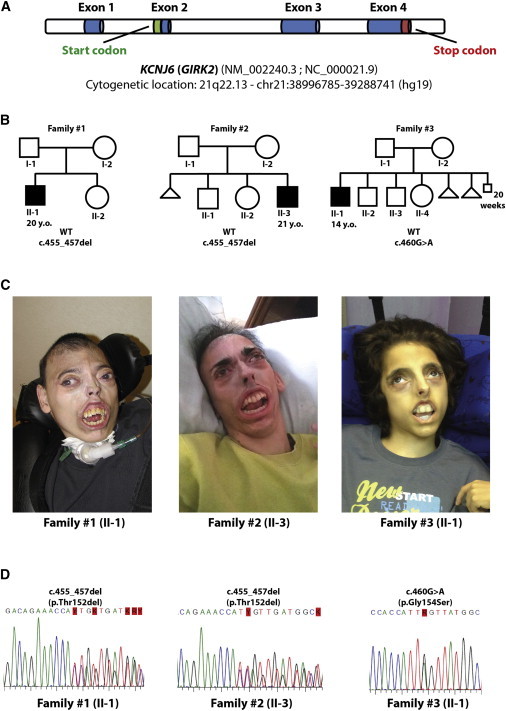
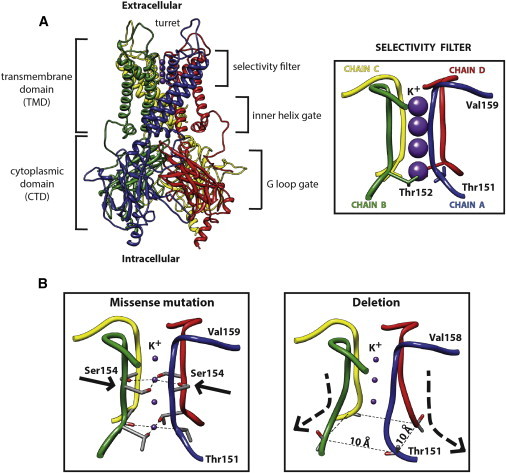
Keppen-Lubinsky syndrome (KPLBS) is a rare disease mainly characterized by severe developmental delay and intellectual disability, microcephaly, large prominent eyes, a narrow nasal bridge, a tented upper lip, a high palate, an open mouth, tightly adherent skin, an aged appearance, and severe generalized lipodystrophy. We sequenced the exomes of three unrelated individuals affected by KPLBS and found de novo heterozygous mutations in KCNJ6 (GIRK2), which encodes an inwardly rectifying potassium channel and maps to the Down syndrome critical region between DIRK1A and DSCR4. In particular, two individuals shared an in-frame heterozygous deletion of three nucleotides (c.455_457del) leading to the loss of one amino acid (p.Thr152del). The third individual was heterozygous for a missense mutation (c.460G>A) which introduces an amino acid change from glycine to serine (p.Gly154Ser). In agreement with animal models, the present data suggest that these mutations severely impair the correct functioning of this potassium channel. Overall, these results establish KPLBS as a channelopathy and suggest that KCNJ6 (GIRK2) could also be a candidate gene for other lipodystrophies. We hope that these results will prompt investigations in this unexplored class of inwardly rectifying K(+) channels.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Gorlin R.J., Cohen M.M., Hennekam R.C.M. Oxford University Press: Oxford; 2001. Syndromes of the Head and Neck.
-
- De Brasi D., Brunetti-Pierri N., Di Micco P., Andria G., Sebastio G. New syndrome with generalized lipodystrophy and a distinctive facial appearance: confirmation of Keppen-Lubinski syndrome? Am. J. Med. Genet. A. 2003;117A:194–195. - PubMed
-
- Basel-Vanagaite L., Shaffer L., Chitayat D. Keppen-Lubinsky syndrome: Expanding the phenotype. Am. J. Med. Genet. A. 2009;149A:1827–1829. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
