

A novel one-class SVM based negative data sampling method for reconstructing proteome-wide HTLV-human protein interaction networks
- PMID: 25620466
- PMCID: PMC5379509
- DOI: 10.1038/srep08034
A novel one-class SVM based negative data sampling method for reconstructing proteome-wide HTLV-human protein interaction networks
Abstract
Protein-protein interaction (PPI) prediction is generally treated as a problem of binary classification wherein negative data sampling is still an open problem to be addressed. The commonly used random sampling is prone to yield less representative negative data with considerable false negatives. Meanwhile rational constraints are seldom exerted on model selection to reduce the risk of false positive predictions for most of the existing computational methods. In this work, we propose a novel negative data sampling method based on one-class SVM (support vector machine, SVM) to predict proteome-wide protein interactions between HTLV retrovirus and Homo sapiens, wherein one-class SVM is used to choose reliable and representative negative data, and two-class SVM is used to yield proteome-wide outcomes as predictive feedback for rational model selection. Computational results suggest that one-class SVM is more suited to be used as negative data sampling method than two-class PPI predictor, and the predictive feedback constrained model selection helps to yield a rational predictive model that reduces the risk of false positive predictions. Some predictions have been validated by the recent literature. Lastly, gene ontology based clustering of the predicted PPI networks is conducted to provide valuable cues for the pathogenesis of HTLV retrovirus.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
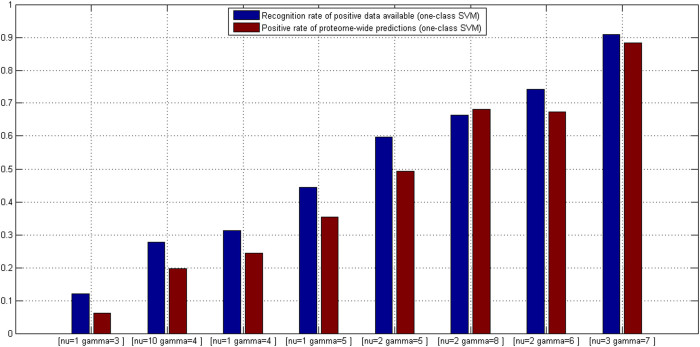
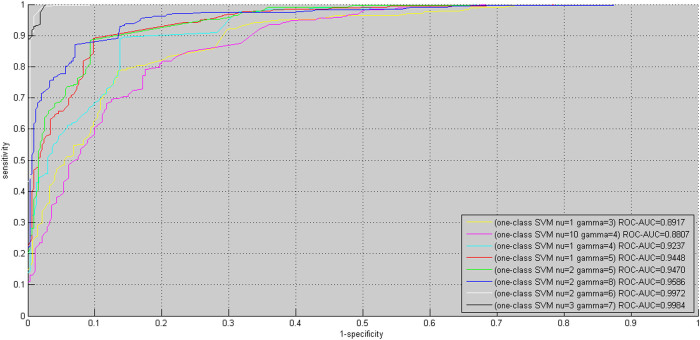
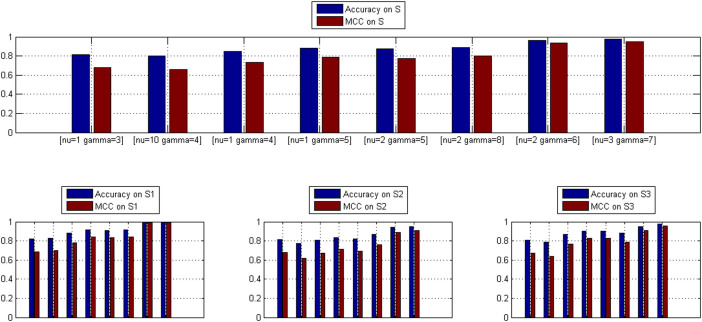
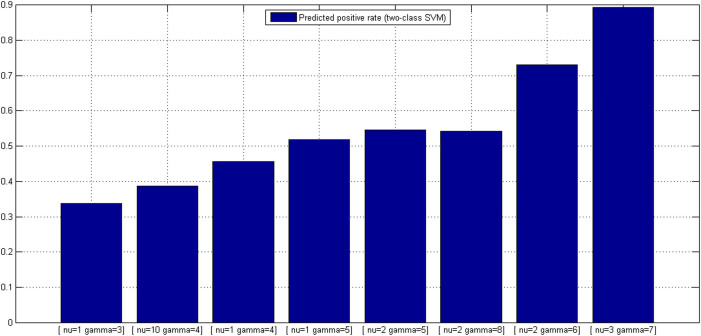
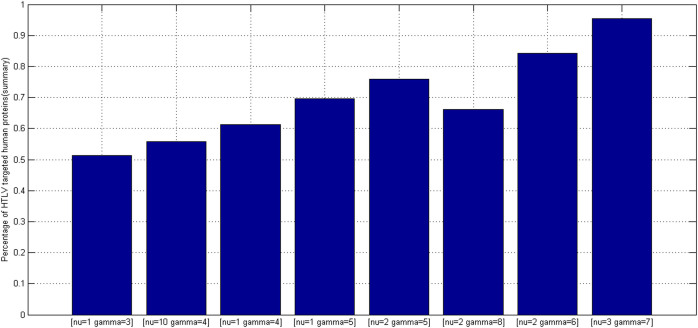
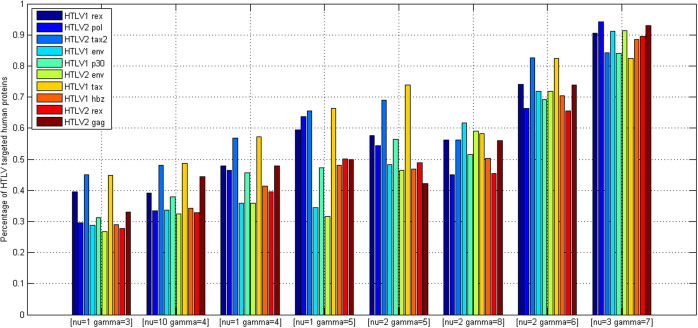
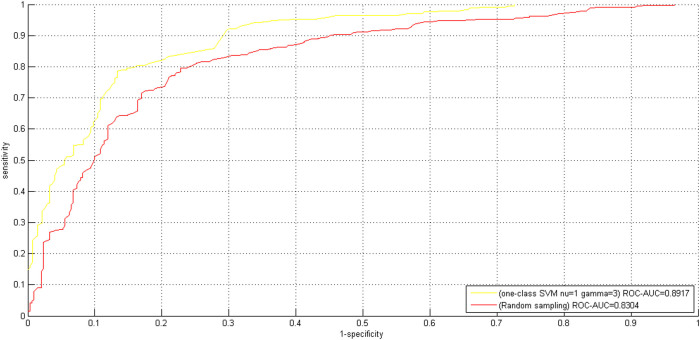
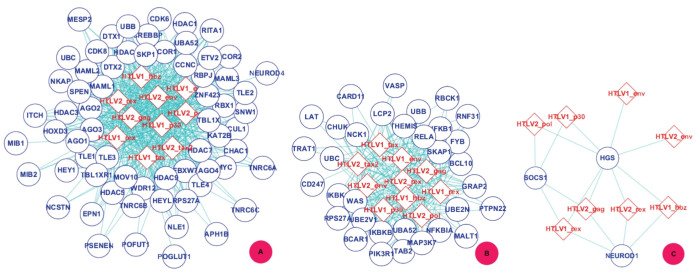
 GO:0007219 - Notch signaling pathway.
GO:0007219 - Notch signaling pathway.  GO:0050852 - T cell receptor signaling pathway.
GO:0050852 - T cell receptor signaling pathway.  GO:0046426 - negative regulation of JAK-STAT cascade. The diamond denotes HTLV proteins and the ecllipse circle denotes human proteins.
GO:0046426 - negative regulation of JAK-STAT cascade. The diamond denotes HTLV proteins and the ecllipse circle denotes human proteins.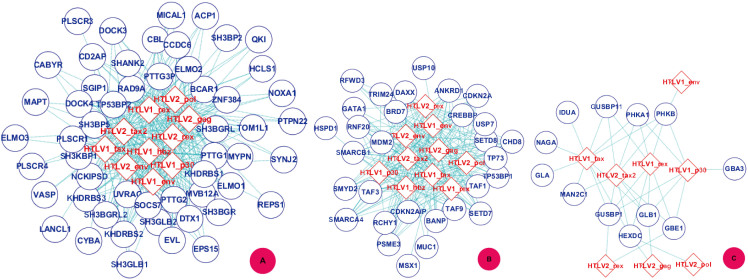
 GO:0017124 - SH3 domain binding.
GO:0017124 - SH3 domain binding.  GO:0002039 - p53 binding.
GO:0002039 - p53 binding.  GO:0004553 - hydrolase activity. The diamond denotes HTLV proteins and the ecllipse circle denotes human proteins.
GO:0004553 - hydrolase activity. The diamond denotes HTLV proteins and the ecllipse circle denotes human proteins.Similar articles
-
Computational reconstruction of proteome-wide protein interaction networks between HTLV retroviruses and Homo sapiens.BMC Bioinformatics. 2014 Jul 18;15(1):245. doi: 10.1186/1471-2105-15-245. BMC Bioinformatics. 2014. PMID: 25037487 Free PMC article.
-
AdaBoost based multi-instance transfer learning for predicting proteome-wide interactions between Salmonella and human proteins.PLoS One. 2014 Oct 17;9(10):e110488. doi: 10.1371/journal.pone.0110488. eCollection 2014. PLoS One. 2014. PMID: 25330226 Free PMC article.
-
Improved Prediction of Protein-Protein Interaction Mapping on Homo Sapiens by Using Amino Acid Sequence Features in a Supervised Learning Framework.Protein Pept Lett. 2021;28(1):74-83. doi: 10.2174/0929866527666200610141258. Protein Pept Lett. 2021. PMID: 32520672
-
Exploring the relationship between hub proteins and drug targets based on GO and intrinsic disorder.Comput Biol Chem. 2015 Jun;56:41-8. doi: 10.1016/j.compbiolchem.2015.03.003. Epub 2015 Mar 23. Comput Biol Chem. 2015. PMID: 25854804
-
Machine-learning techniques for the prediction of protein-protein interactions.J Biosci. 2019 Sep;44(4):104. J Biosci. 2019. PMID: 31502581
Cited by
-
Multi-label ℓ2-regularized logistic regression for predicting activation/inhibition relationships in human protein-protein interaction networks.Sci Rep. 2016 Nov 7;6:36453. doi: 10.1038/srep36453. Sci Rep. 2016. PMID: 27819359 Free PMC article.
-
Assisting document triage for human kinome curation via machine learning.Database (Oxford). 2018 Jan 1;2018:bay091. doi: 10.1093/database/bay091. Database (Oxford). 2018. PMID: 30239677 Free PMC article.
-
APBIO: bioactive profiling of air pollutants through inferred bioactivity signatures and prediction of novel target interactions.J Cheminform. 2025 Jan 31;17(1):13. doi: 10.1186/s13321-025-00961-1. J Cheminform. 2025. PMID: 39891207 Free PMC article.
-
A computational framework for distinguishing direct versus indirect interactions in human functional protein-protein interaction networks.Integr Biol (Camb). 2017 Jul 17;9(7):595-606. doi: 10.1039/c7ib00013h. Integr Biol (Camb). 2017. PMID: 28524201 Free PMC article.
-
Uncovering New Pathogen-Host Protein-Protein Interactions by Pairwise Structure Similarity.PLoS One. 2016 Jan 22;11(1):e0147612. doi: 10.1371/journal.pone.0147612. eCollection 2016. PLoS One. 2016. PMID: 26799490 Free PMC article.
References
-
- Jansen R., Gerstein M. Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Curr Opin Microbiol 7, 535–545 (2004). - PubMed
-
- Dyer M., Murali T., Sobral B. Computational prediction of host-pathogen protein-protein interactions. Bioinformatics 23, i159–i166 (2007). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
