

Reconstructing each cell's genome within complex microbial communities-dream or reality?
- PMID: 25620966
- PMCID: PMC4287102
- DOI: 10.3389/fmicb.2014.00771
Reconstructing each cell's genome within complex microbial communities-dream or reality?
Abstract
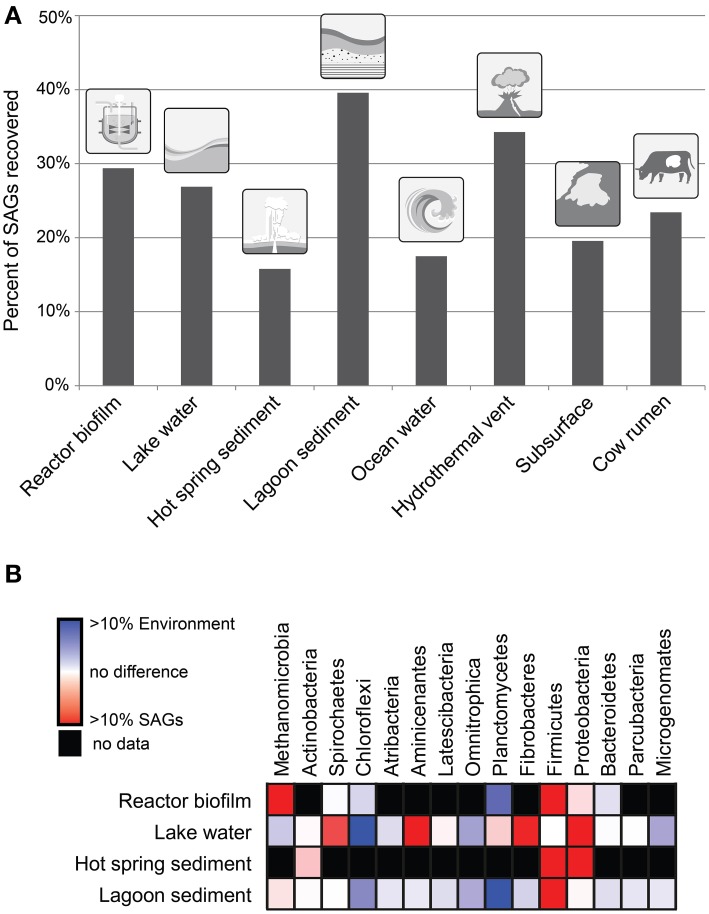
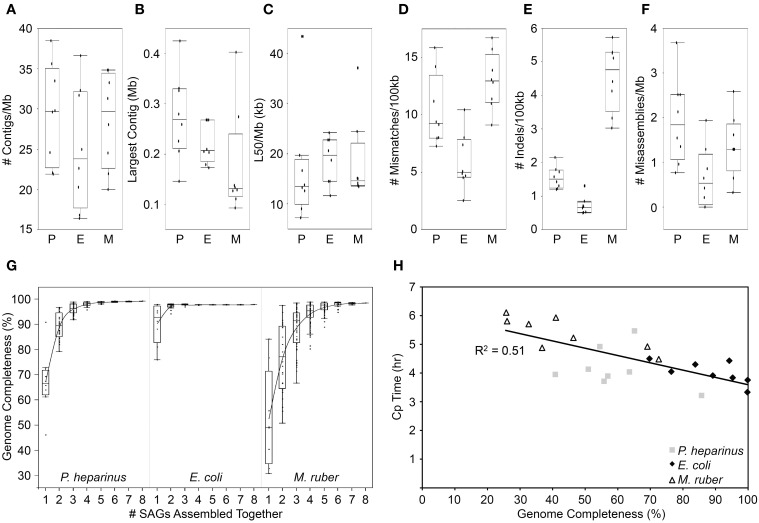
As the vast majority of microorganisms have yet to be cultivated in a laboratory setting, access to their genetic makeup has largely been limited to cultivation-independent methods. These methods, namely metagenomics and more recently single-cell genomics, have become cornerstones for microbial ecology and environmental microbiology. One ultimate goal is the recovery of genome sequences from each cell within an environment to move toward a better understanding of community metabolic potential and to provide substrate for experimental work. As single-cell sequencing has the ability to decipher all sequence information contained in an individual cell, this method holds great promise in tackling such challenge. Methodological limitations and inherent biases however do exist, which will be discussed here based on environmental and benchmark data, to assess how far we are from reaching this goal.
Keywords: environmental microbiology; genome completeness; genome quality; microbial ecology; multiple displacement amplification; single-cell sequencing.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials