

Selective impairment of methylation maintenance is the major cause of DNA methylation reprogramming in the early embryo
- PMID: 25621012
- PMCID: PMC4304184
- DOI: 10.1186/1756-8935-8-1
Selective impairment of methylation maintenance is the major cause of DNA methylation reprogramming in the early embryo
Abstract
Background: DNA methylomes are extensively reprogrammed during mouse pre-implantation and early germ cell development. The main feature of this reprogramming is a genome-wide decrease in 5-methylcytosine (5mC). Standard high-resolution single-stranded bisulfite sequencing techniques do not allow discrimination of the underlying passive (replication-dependent) or active enzymatic mechanisms of 5mC loss. We approached this problem by generating high-resolution deep hairpin bisulfite sequencing (DHBS) maps, allowing us to follow the patterns of symmetric DNA methylation at CpGs dyads on both DNA strands over single replications.
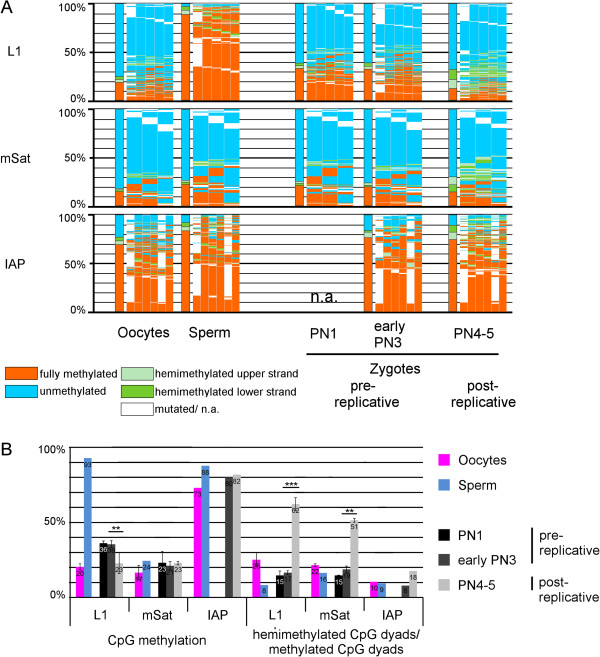
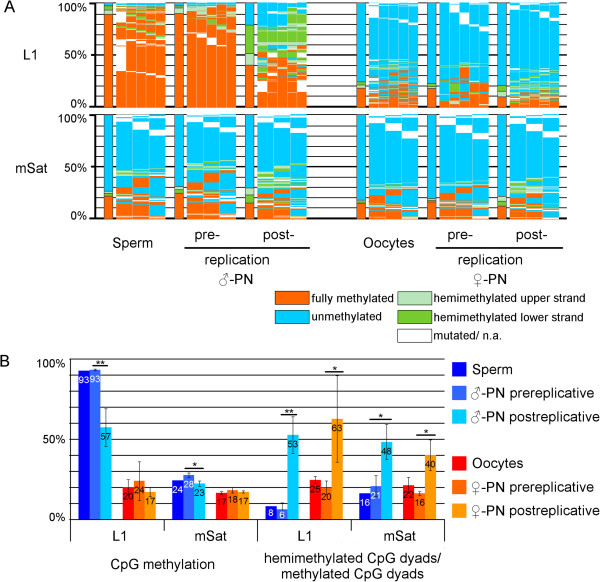
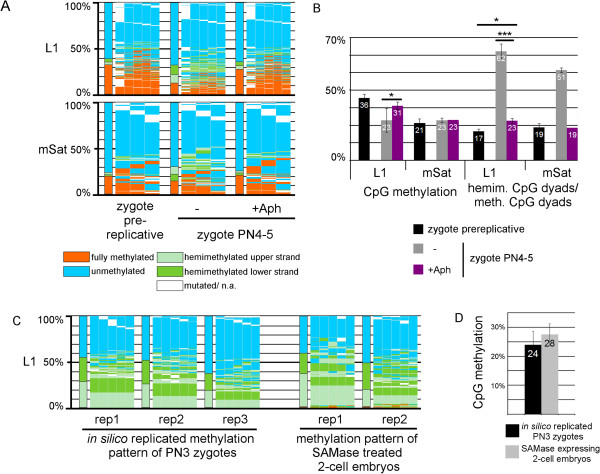
Results: We compared DHBS maps of repetitive elements in the developing zygote, the early embryo, and primordial germ cells (PGCs) at defined stages of development. In the zygote, we observed distinct effects in paternal and maternal chromosomes. A significant loss of paternal DNA methylation was linked to replication and to an increase in continuous and dispersed hemimethylated CpG dyad patterns. Overall methylation levels at maternal copies remained largely unchanged, but showed an increased level of dispersed hemi-methylated CpG dyads. After the first cell cycle, the combined DHBS patterns of paternal and maternal chromosomes remained unchanged over the next three cell divisions. By contrast, in PGCs the DNA demethylation process was continuous, as seen by a consistent decrease in fully methylated CpG dyads over consecutive cell divisions.
Conclusions: The main driver of DNA demethylation in germ cells and in the zygote is partial impairment of maintenance of symmetric DNA methylation at CpG dyads. In the embryo, this passive demethylation is restricted to the first cell division, whereas it continues over several cell divisions in germ cells. The dispersed patterns of CpG dyads in the early-cleavage embryo suggest a continuous partial (and to a low extent active) loss of methylation apparently compensated for by selective de novo methylation. We conclude that a combination of passive and active demethylation events counteracted by de novo methylation are involved in the distinct reprogramming dynamics of DNA methylomes in the zygote, the early embryo, and PGCs.
Keywords: DNA methylation pattern; DNA methylation reprogramming; Deep hairpin bisulfite sequencing; Pre-implantation development.
Figures



References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
