

The Use of Patient-Specific Induced Pluripotent Stem Cells (iPSCs) to Identify Osteoclast Defects in Rare Genetic Bone Disorders
- PMID: 25621177
- PMCID: PMC4300535
- DOI: 10.3390/jcm3041490
The Use of Patient-Specific Induced Pluripotent Stem Cells (iPSCs) to Identify Osteoclast Defects in Rare Genetic Bone Disorders
Abstract
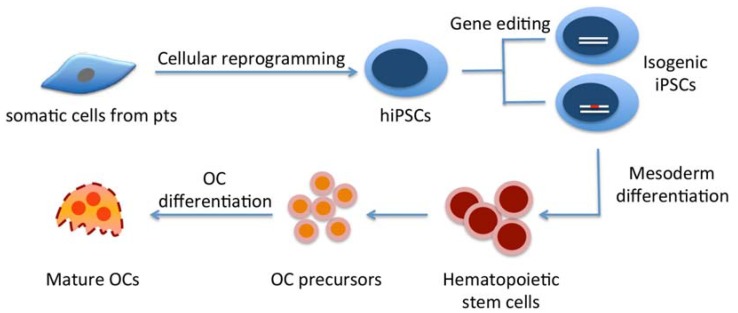
More than 500 rare genetic bone disorders have been described, but for many of them only limited treatment options are available. Challenges for studying these bone diseases come from a lack of suitable animal models and unavailability of skeletal tissues for studies. Effectors for skeletal abnormalities of bone disorders may be abnormal bone formation directed by osteoblasts or anomalous bone resorption by osteoclasts, or both. Patient-specific induced pluripotent stem cells (iPSCs) can be generated from somatic cells of various tissue sources and in theory can be differentiated into any desired cell type. However, successful differentiation of hiPSCs into functional bone cells is still a challenge. Our group focuses on the use of human iPSCs (hiPSCs) to identify osteoclast defects in craniometaphyseal dysplasia. In this review, we describe the impact of stem cell technology on research for better treatment of such disorders, the generation of hiPSCs from patients with rare genetic bone disorders and current protocols for differentiating hiPSCs into osteoclasts.
Keywords: induced pluripotent stem cells; osteoclast; rare genetic bone disorders.
Figures
Similar articles
-
Differentiation of Human Induced Pluripotent Stem Cells (hiPSCs) into Osteoclasts.Bio Protoc. 2020 Dec 20;10(24):e3854. doi: 10.21769/BioProtoc.3854. eCollection 2020 Dec 20. Bio Protoc. 2020. PMID: 33659501 Free PMC article.
-
Craniometaphyseal Dysplasia Mutations in ANKH Negatively Affect Human Induced Pluripotent Stem Cell Differentiation into Osteoclasts.Stem Cell Reports. 2017 Nov 14;9(5):1369-1376. doi: 10.1016/j.stemcr.2017.09.016. Epub 2017 Oct 19. Stem Cell Reports. 2017. PMID: 29056330 Free PMC article.
-
Targeted reversion of induced pluripotent stem cells from patients with human cleidocranial dysplasia improves bone regeneration in a rat calvarial bone defect model.Stem Cell Res Ther. 2018 Jan 22;9(1):12. doi: 10.1186/s13287-017-0754-4. Stem Cell Res Ther. 2018. PMID: 29357927 Free PMC article.
-
Human-Induced Pluripotent Stem Cells in Plastic and Reconstructive Surgery.Int J Mol Sci. 2024 Feb 3;25(3):1863. doi: 10.3390/ijms25031863. Int J Mol Sci. 2024. PMID: 38339142 Free PMC article. Review.
-
Concise review: embryonic stem cells: a new tool to study osteoblast and osteoclast differentiation.Stem Cells. 2007 Mar;25(3):544-52. doi: 10.1634/stemcells.2006-0395. Epub 2006 Nov 9. Stem Cells. 2007. PMID: 17095705 Review.
Cited by
-
Comparison of osteoclast differentiation protocols from human induced pluripotent stem cells of different tissue origins.Stem Cell Res Ther. 2023 Nov 7;14(1):319. doi: 10.1186/s13287-023-03547-6. Stem Cell Res Ther. 2023. PMID: 37936199 Free PMC article.
-
Differentiation of Human Induced Pluripotent Stem Cells (hiPSCs) into Osteoclasts.Bio Protoc. 2020 Dec 20;10(24):e3854. doi: 10.21769/BioProtoc.3854. eCollection 2020 Dec 20. Bio Protoc. 2020. PMID: 33659501 Free PMC article.
-
Mesenchymal Stem Cell Therapy for Bone Regeneration.Clin Orthop Surg. 2018 Sep;10(3):271-278. doi: 10.4055/cios.2018.10.3.271. Epub 2018 Aug 22. Clin Orthop Surg. 2018. PMID: 30174801 Free PMC article. Review.
-
Induced pluripotent stem cell technology in bone biology.Bone. 2023 Jul;172:116760. doi: 10.1016/j.bone.2023.116760. Epub 2023 Apr 6. Bone. 2023. PMID: 37028583 Free PMC article.
-
Bone Cells Differentiation: How CFTR Mutations May Rule the Game of Stem Cells Commitment?Front Cell Dev Biol. 2021 May 7;9:611921. doi: 10.3389/fcell.2021.611921. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34026749 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources