

Plasticity of DNA methylation in a nerve injury model of pain
- PMID: 25621511
- PMCID: PMC4622653
- DOI: 10.1080/15592294.2015.1006493
Plasticity of DNA methylation in a nerve injury model of pain
Abstract
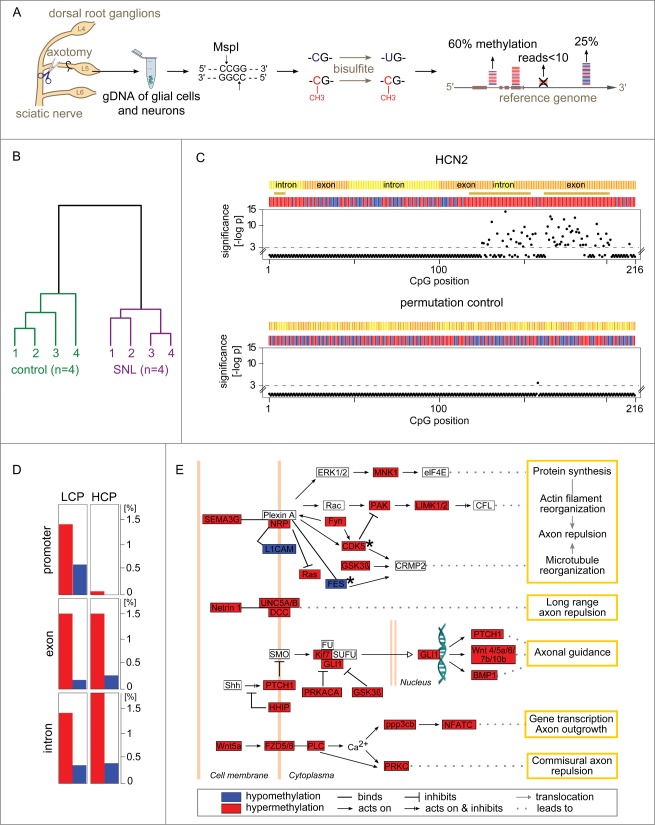
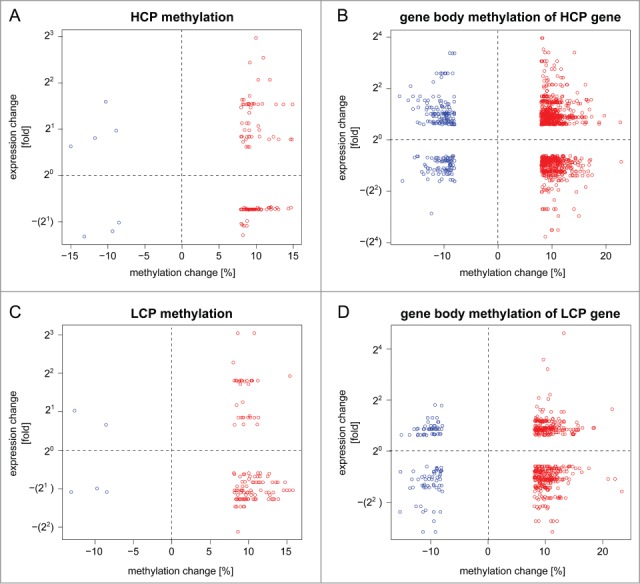
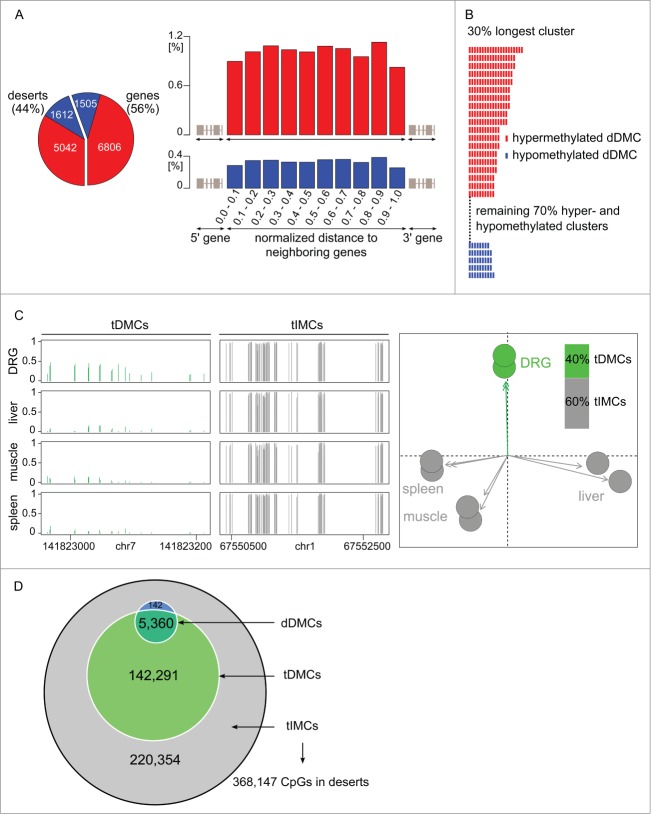
The response of the peripheral nervous system (PNS) to injury may go together with alterations in epigenetics, a conjecture that has not been subjected to a comprehensive, genome-wide test. Using reduced representation bisulfite sequencing, we report widespread remodeling of DNA methylation in the rat dorsal root ganglion (DRG) occurring within 24 h of peripheral nerve ligation, a neuropathy model of allodynia. Significant (P < 10(-4)) cytosine hyper- and hypo-methylation was found at thousands of CpG sites. Remodeling occurred outside of CpG islands. Changes affected genes with known roles in the PNS, yet methylome remodeling also involved genes that were not linked to neuroplasticity by prior evidence. Consistent with emerging models relying on genome-wide methylation and RNA-seq analysis of promoter regions and gene bodies, variation of methylation was not tightly linked with variation of gene expression. Furthermore, approximately 44% of the dynamically changed CpGs were located outside of genes. We compared their positions with the intergenic, tissue-specific differentially methylated CpGs (tDMCs) of an independent experimental set consisting of liver, spleen, L4 control DRG, and muscle. Dynamic changes affected those intergenic CpGs that were different between tissues (P < 10(-15)) and almost never the invariant portion of the methylome (those CpGs that were identical across all tissues). Our findings-obtained in mixed tissue-show that peripheral nerve injury leads to methylome remodeling in the DRG. Future studies may address which of the cell types found in the DRG, such as specific groups of neurons or non-neuronal cells are affected by which aspect of the observed methylome remodeling.
Keywords: DRG, dorsal root ganglion; HCP, high-CpG promoter; LCP, low-CpG promoter; RRBS; RRBS, reduced representation bisulfite sequencing; SNL, spinal nerve ligation; dDMCs, dynamically differentially methylated CpGs; dorsal root ganglion; methylation; pain; peripheral nervous system; rat; spinal nerve ligation; tDMCs, tissue-specific differentially methylated CpGs; tIMCs, tissue-invariant methylated CpGs.
Figures
Similar articles
-
DNA Methylation and Non-Coding RNAs during Tissue-Injury Associated Pain.Int J Mol Sci. 2022 Jan 11;23(2):752. doi: 10.3390/ijms23020752. Int J Mol Sci. 2022. PMID: 35054943 Free PMC article. Review.
-
Nerve Injury-Induced Chronic Pain Is Associated with Persistent DNA Methylation Reprogramming in Dorsal Root Ganglion.J Neurosci. 2018 Jul 4;38(27):6090-6101. doi: 10.1523/JNEUROSCI.2616-17.2018. Epub 2018 Jun 6. J Neurosci. 2018. PMID: 29875269 Free PMC article.
-
Diametrically opposite methylome-transcriptome relationships in high- and low-CpG promoter genes in postmitotic neural rat tissue.Epigenetics. 2012 May;7(5):421-8. doi: 10.4161/epi.19565. Epub 2012 May 1. Epigenetics. 2012. PMID: 22415013 Free PMC article.
-
Increased expression of HCN2 channel protein in L4 dorsal root ganglion neurons following axotomy of L5- and inflammation of L4-spinal nerves in rats.Neuroscience. 2015 Jun 4;295:90-102. doi: 10.1016/j.neuroscience.2015.03.041. Epub 2015 Mar 24. Neuroscience. 2015. PMID: 25813712
-
Genome-scale DNA methylation analysis.Epigenomics. 2010 Feb;2(1):105-17. doi: 10.2217/epi.09.35. Epigenomics. 2010. PMID: 20657796 Free PMC article. Review.
Cited by
-
Epigenetic Landscapes of Pain: DNA Methylation Dynamics in Chronic Pain.Int J Mol Sci. 2024 Jul 30;25(15):8324. doi: 10.3390/ijms25158324. Int J Mol Sci. 2024. PMID: 39125894 Free PMC article. Review.
-
DNA Methylation and Non-Coding RNAs during Tissue-Injury Associated Pain.Int J Mol Sci. 2022 Jan 11;23(2):752. doi: 10.3390/ijms23020752. Int J Mol Sci. 2022. PMID: 35054943 Free PMC article. Review.
-
The transition from acute to chronic pain: dynamic epigenetic reprogramming of the mouse prefrontal cortex up to 1 year after nerve injury.Pain. 2020 Oct;161(10):2394-2409. doi: 10.1097/j.pain.0000000000001917. Pain. 2020. PMID: 32427748 Free PMC article.
-
Genome-wide redistribution of MeCP2 in dorsal root ganglia after peripheral nerve injury.Epigenetics Chromatin. 2016 Jun 7;9:23. doi: 10.1186/s13072-016-0073-5. eCollection 2016. Epigenetics Chromatin. 2016. PMID: 27279901 Free PMC article.
-
Etiology matters - Genomic DNA Methylation Patterns in Three Rat Models of Acquired Epilepsy.Sci Rep. 2016 May 9;6:25668. doi: 10.1038/srep25668. Sci Rep. 2016. PMID: 27157830 Free PMC article.
References
-
- Costigan M, Belfer I, Griffin RS, Dai F, Barrett LB, Coppola G, Wu T, Kiselycznyk C, Poddar M, Lu Y, et al. . Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain 2010; 133:2519-27; PMID:20724292; http://dx.doi.org/10.1093/brain/awq195 - DOI - PMC - PubMed
-
- Hammer P, Banck MS, Amberg R, Wang C, Petznick G, Luo S, Khrebtukova I, Schroth GP, Beyerlein P, Beutler AS. mRNA-seq with agnostic splice site discovery for nervous system transcriptomics tested in chronic pain. Genome Res 2010; 20:847-60; PMID:20452967; http://dx.doi.org/10.1101/gr.101204.109 - DOI - PMC - PubMed
-
- Michaelevski I, Segal-Ruder Y, Rozenbaum M, Medzihradszky KF, Shalem O, Coppola G, Horn-Saban S, Ben-Yaakov K, Dagan SY, Rishal I, et al. . Signaling to transcription networks in the neuronal retrograde injury response. Sci Signal 2010; 3:ra53; PMID:20628157; http://dx.doi.org/10.1126/scisignal.2000952 - DOI - PMC - PubMed
-
- Zhang Z, Cai Y-Q, Zou F, Bie B, Pan ZZ. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med 2011; 17:1448-55; PMID:21983856; http://dx.doi.org/10.1038/nm.2442 - DOI - PMC - PubMed
-
- Tajerian M, Alvarado S, Millecamps M, Dashwood T, Anderson KM, Haglund L, Ouellet J, Szyf M, Stone LS. DNA methylation of SPARC and chronic low back pain. Mol Pain 2011; 7:65; PMID:21867537; http://dx.doi.org/10.1186/1744-8069-7-65 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous