

Lagging-strand replication shapes the mutational landscape of the genome
- PMID: 25624100
- PMCID: PMC4374164
- DOI: 10.1038/nature14183
Lagging-strand replication shapes the mutational landscape of the genome
Abstract
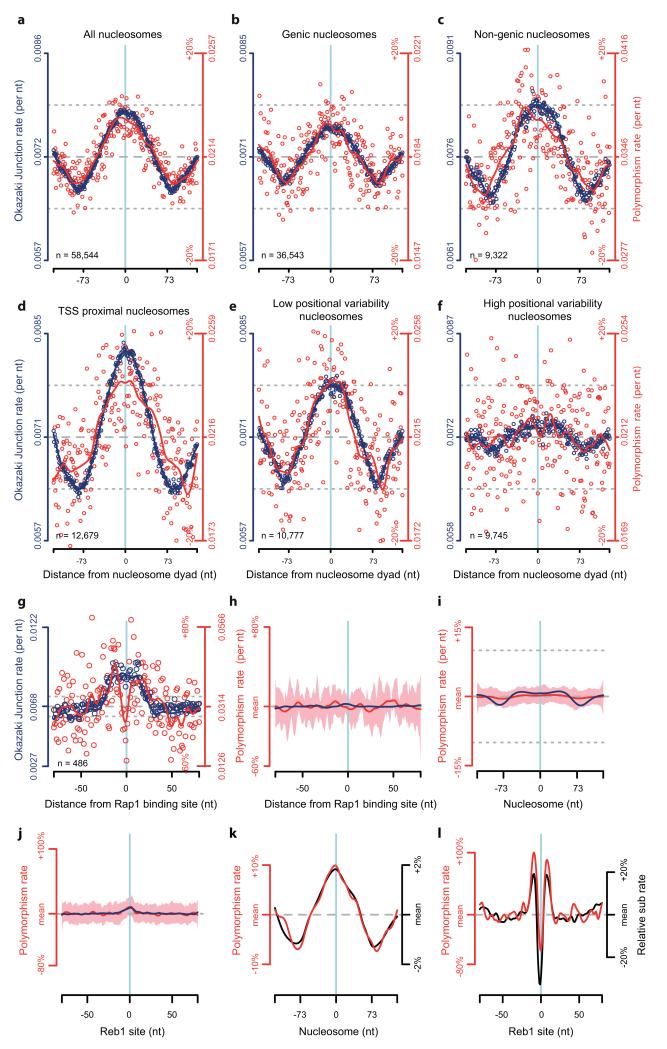
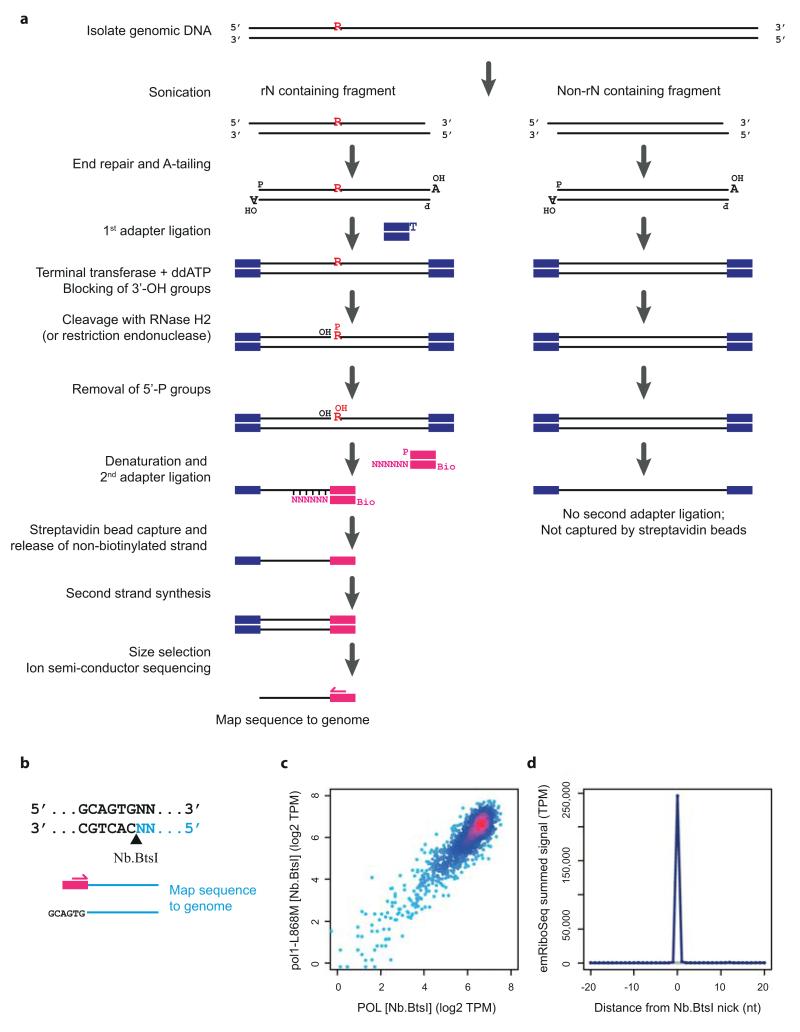
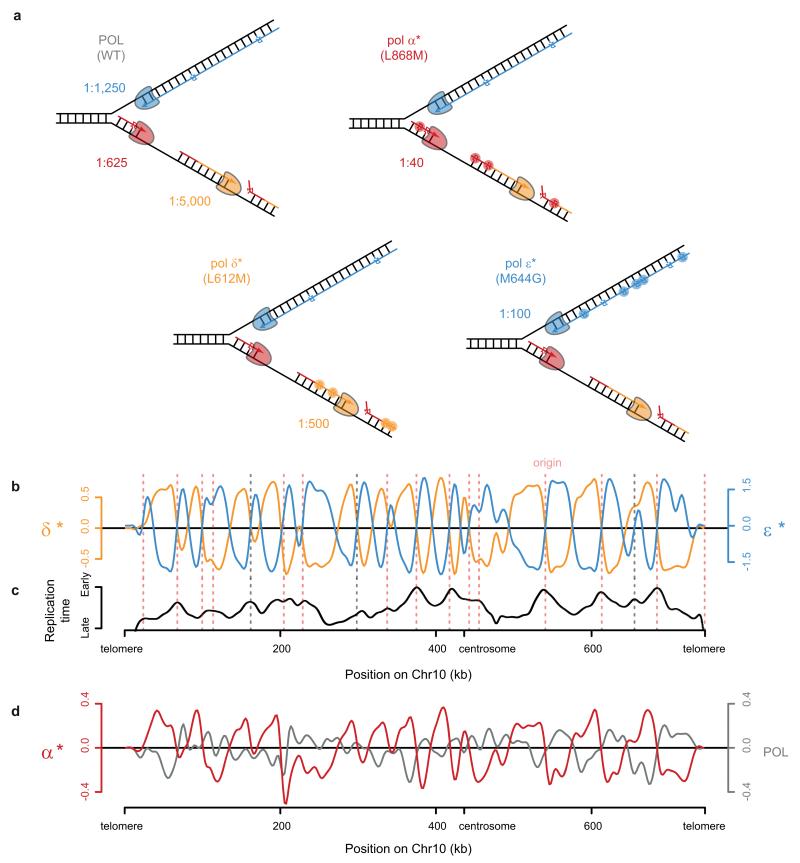
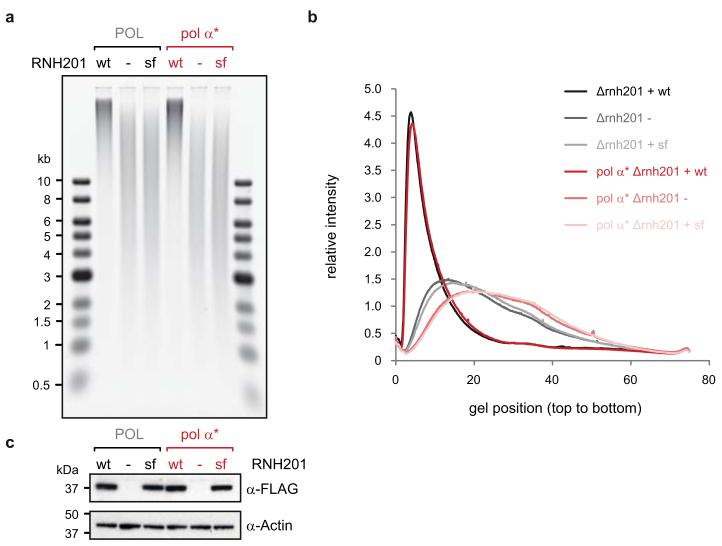
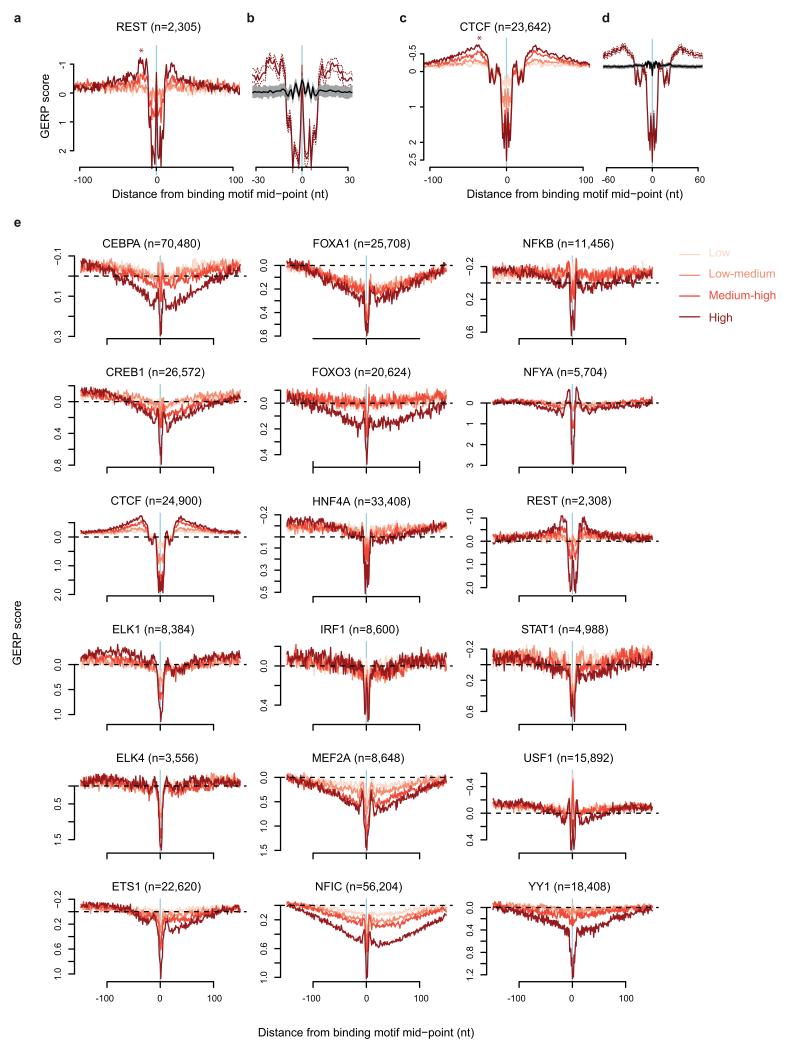
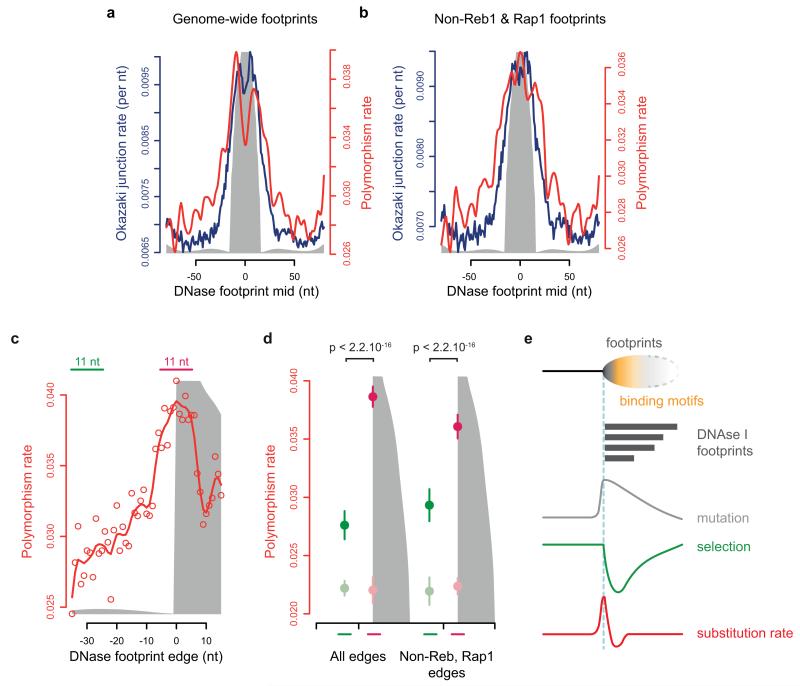
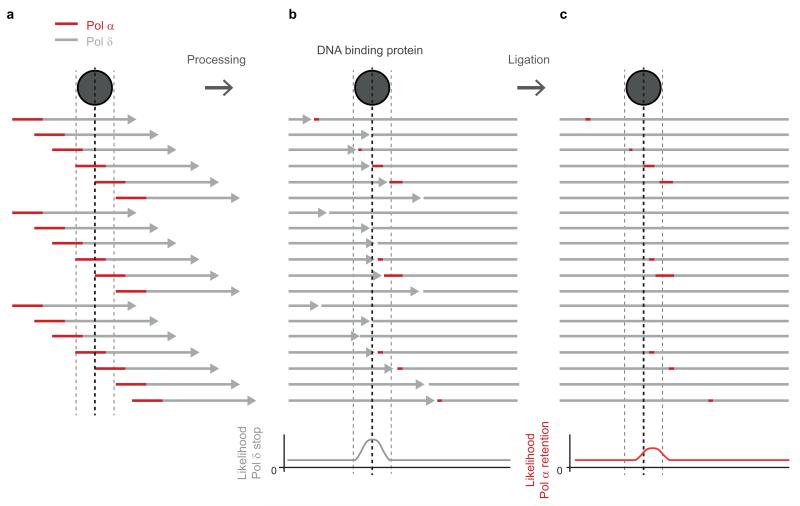
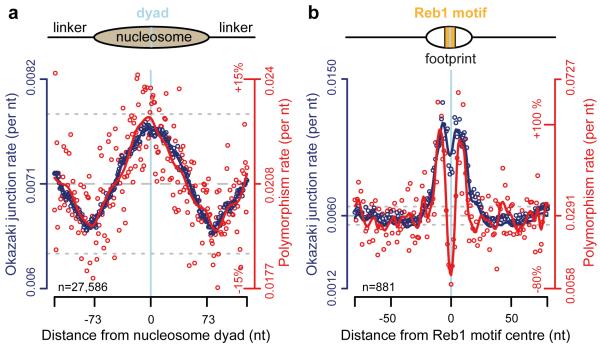
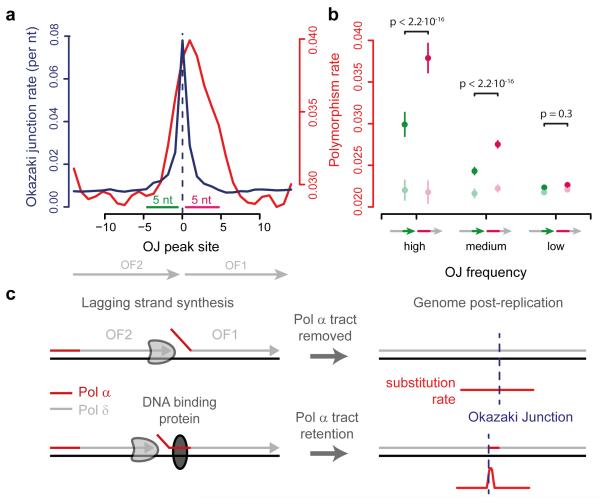
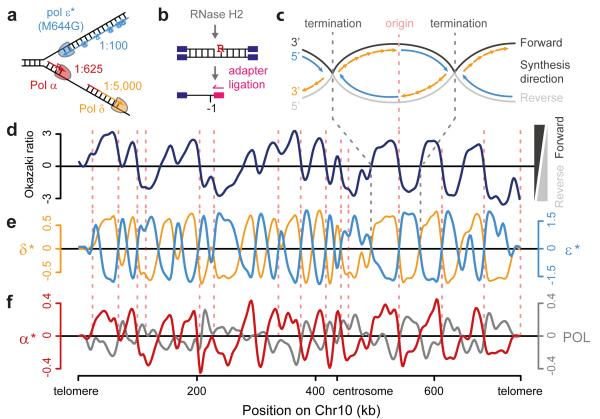
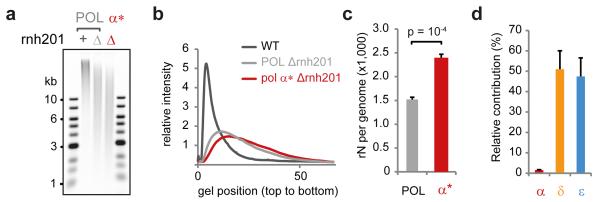
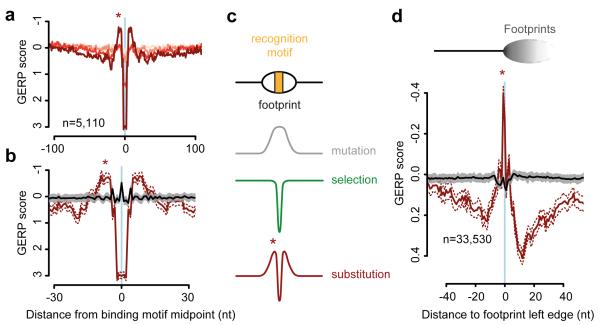
The origin of mutations is central to understanding evolution and of key relevance to health. Variation occurs non-randomly across the genome, and mechanisms for this remain to be defined. Here we report that the 5' ends of Okazaki fragments have significantly increased levels of nucleotide substitution, indicating a replicative origin for such mutations. Using a novel method, emRiboSeq, we map the genome-wide contribution of polymerases, and show that despite Okazaki fragment processing, DNA synthesized by error-prone polymerase-α (Pol-α) is retained in vivo, comprising approximately 1.5% of the mature genome. We propose that DNA-binding proteins that rapidly re-associate post-replication act as partial barriers to Pol-δ-mediated displacement of Pol-α-synthesized DNA, resulting in incorporation of such Pol-α tracts and increased mutation rates at specific sites. We observe a mutational cost to chromatin and regulatory protein binding, resulting in mutation hotspots at regulatory elements, with signatures of this process detectable in both yeast and humans.
Figures

References
-
- Wolfe KH, Sharp PM, Li WH. Mutation rates differ among regions of the mammalian genome. Nature. 1989;337:283–5. - PubMed
Methods References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
