

NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases
- PMID: 25625584
- PMCID: PMC4315937
- DOI: 10.1016/j.redox.2015.01.008
NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases
Abstract
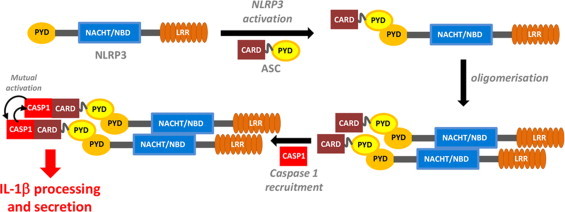
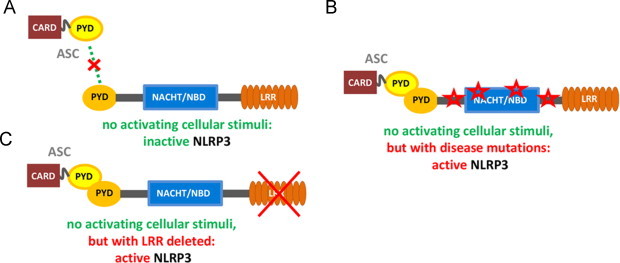
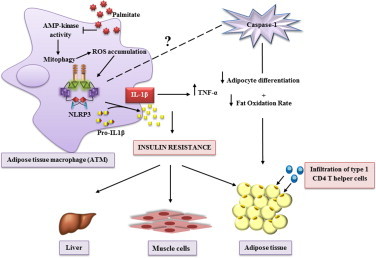
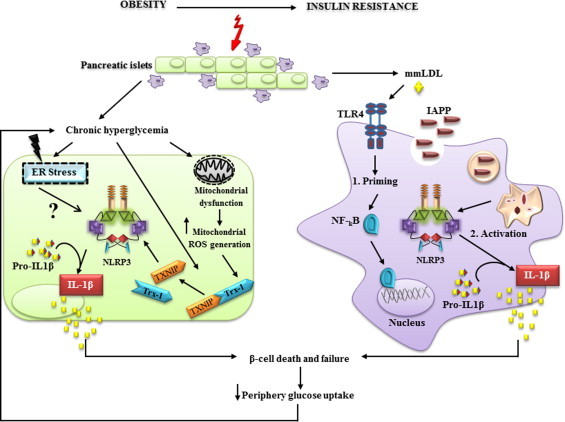
IL-1β production is critically regulated by cytosolic molecular complexes, termed inflammasomes. Different inflammasome complexes have been described to date. While all inflammasomes recognize certain pathogens, it is the distinctive feature of NLRP3 inflammasome to be activated by many and diverse stimuli making NLRP3 the most versatile, and importantly also the most clinically implicated inflammasome. However, NLRP3 activation has remained the most enigmatic. It is not plausible that the intracellular NLRP3 receptor is able to detect all of its many and diverse triggers through direct interactions; instead, it is discussed that NLRP3 is responding to certain generic cellular stress-signals induced by the multitude of molecules that trigger its activation. An ever increasing number of studies link the sensing of cellular stress signals to a direct pathophysiological role of NLRP3 activation in a wide range of autoinflammatory and autoimmune disorders, and thus provide a novel mechanistic rational, on how molecules trigger and support sterile inflammatory diseases. A vast interest has created to unravel how NLRP3 becomes activated, since mechanistic insight is the prerequisite for a knowledge-based development of therapeutic intervention strategies that specifically target the NLRP3 triggered IL-1β production. In this review, we have updated knowledge on NLRP3 inflammasome assembly and activation and on the pyrin domain in NLRP3 that could represent a drug target to treat sterile inflammatory diseases. We have reported mutations in NLRP3 that were found to be associated with certain diseases. In addition, we have reviewed the functional link between NLRP3 inflammasome, the regulator of cellular redox status Trx/TXNIP complex, endoplasmic reticulum stress and the pathogenesis of diseases such as type 2 diabetes. Finally, we have provided data on NLRP3 inflammasome, as a critical regulator involved in the pathogenesis of obesity and cardiovascular diseases.
Keywords: Cardiovascular diseases; IL-18 (PDB id: Q14116); IL-1β (PDB id: P01584, P10749); NLRP3 inflammasome (PDB id: Q96P20); Obesity.
Copyright © 2015 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
