

Clinical actionability enhanced through deep targeted sequencing of solid tumors
- PMID: 25626406
- PMCID: PMC4511273
- DOI: 10.1373/clinchem.2014.231100
Clinical actionability enhanced through deep targeted sequencing of solid tumors
Abstract
Background: Further advances of targeted cancer therapy require comprehensive in-depth profiling of somatic mutations that are present in subpopulations of tumor cells in a clinical tumor sample. However, it is unclear to what extent such intratumor heterogeneity is present and whether it may affect clinical decision-making. To study this question, we established a deep targeted sequencing platform to identify potentially actionable DNA alterations in tumor samples.
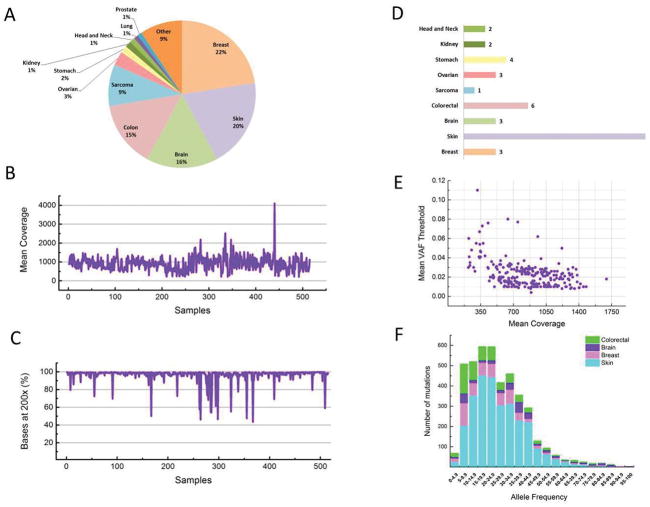
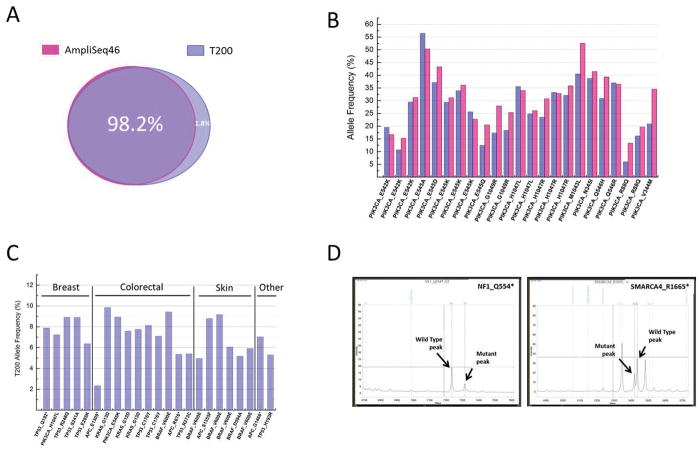
Methods: We assayed 515 formalin-fixed paraffin-embedded (FFPE) tumor samples and matched germline DNA (475 patients) from 11 disease sites by capturing and sequencing all the exons in 201 cancer-related genes. Mutations, indels, and copy number data were reported.
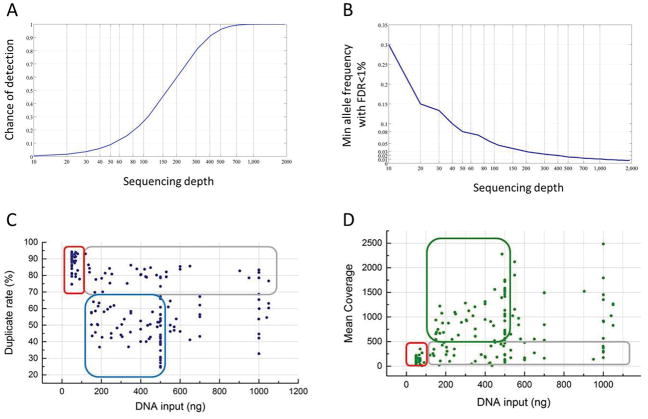
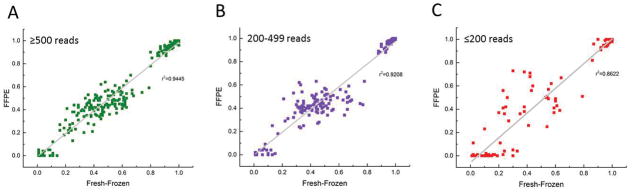
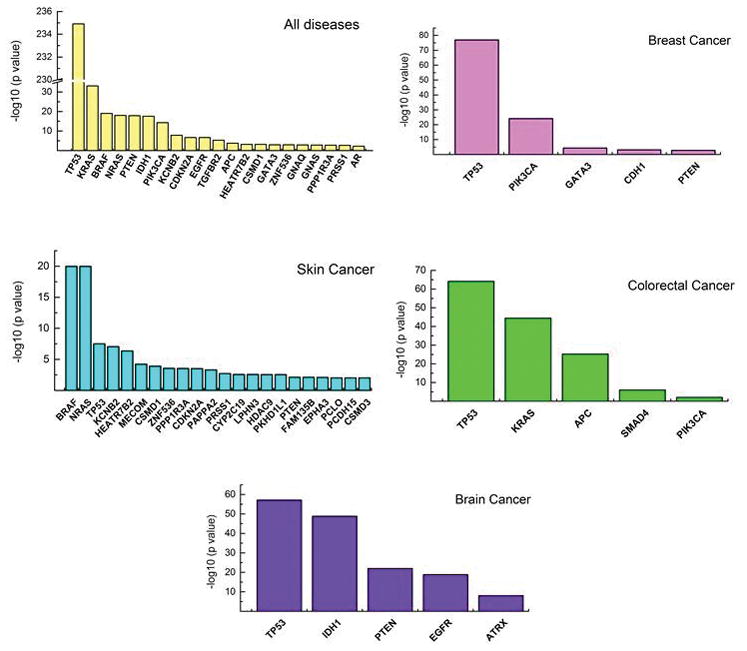
Results: We obtained a 1000-fold mean sequencing depth and identified 4794 nonsynonymous mutations in the samples analyzed, of which 15.2% were present at <10% allele frequency. Most of these low level mutations occurred at known oncogenic hotspots and are likely functional. Identifying low level mutations improved identification of mutations in actionable genes in 118 (24.84%) patients, among which 47 (9.8%) otherwise would have been unactionable. In addition, acquiring ultrahigh depth also ensured a low false discovery rate (<2.2%) from FFPE samples.
Conclusions: Our results were as accurate as a commercially available CLIA-compliant hotspot panel but allowed the detection of a higher number of mutations in actionable genes. Our study reveals the critical importance of acquiring and utilizing high sequencing depth in profiling clinical tumor samples and presents a very useful platform for implementing routine sequencing in a cancer care institution.
© 2014 American Association for Clinical Chemistry.
Conflict of interest statement
GBM: SAB/Consultant: Illumina, AstraZeneca, Blend, Critical Outcome Technologies, HanAl Bio Korea, Nuevolution, Pfizer, Provista Diagnostics, Roche, Signalchem Lifesciences, Symphogen, Tau Therapeutics.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
