

Neuroinflammation and J2 prostaglandins: linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration
- PMID: 25628533
- PMCID: PMC4292445
- DOI: 10.3389/fnmol.2014.00104
Neuroinflammation and J2 prostaglandins: linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration
Abstract
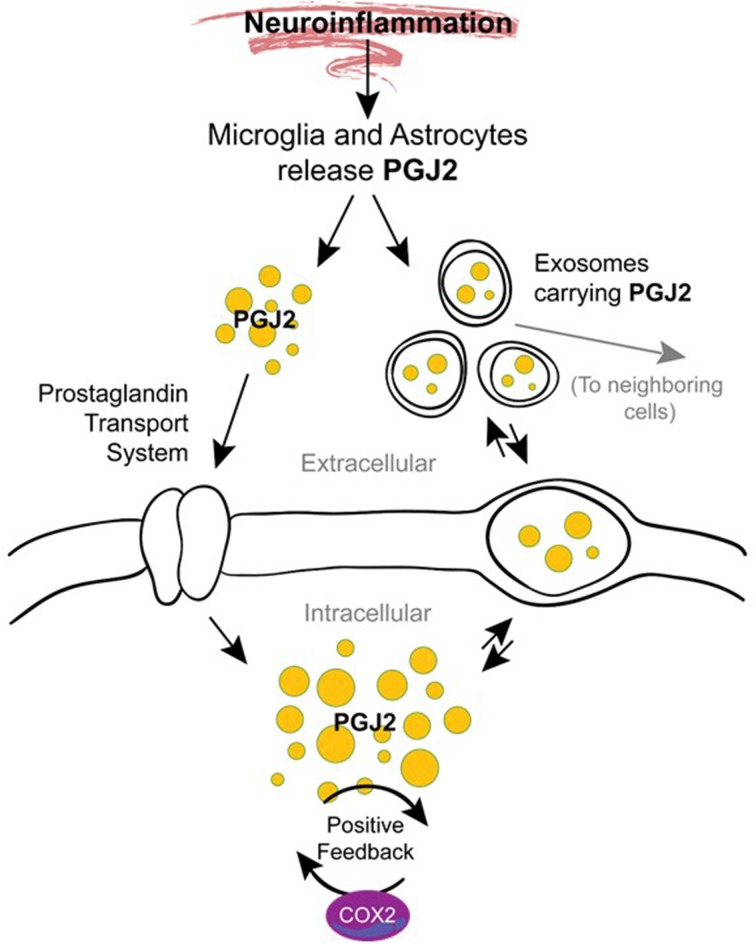
The immune response of the CNS is a defense mechanism activated upon injury to initiate repair mechanisms while chronic over-activation of the CNS immune system (termed neuroinflammation) may exacerbate injury. The latter is implicated in a variety of neurological and neurodegenerative disorders such as Alzheimer and Parkinson diseases, amyotrophic lateral sclerosis, multiple sclerosis, traumatic brain injury, HIV dementia, and prion diseases. Cyclooxygenases (COX-1 and COX-2), which are key enzymes in the conversion of arachidonic acid into bioactive prostanoids, play a central role in the inflammatory cascade. J2 prostaglandins are endogenous toxic products of cyclooxygenases, and because their levels are significantly increased upon brain injury, they are actively involved in neuronal dysfunction induced by pro-inflammatory stimuli. In this review, we highlight the mechanisms by which J2 prostaglandins (1) exert their actions, (2) potentially contribute to the transition from acute to chronic inflammation and to the spreading of neuropathology, (3) disturb the ubiquitin-proteasome pathway and mitochondrial function, and (4) contribute to neurodegenerative disorders such as Alzheimer and Parkinson diseases, and amyotrophic lateral sclerosis, as well as stroke, traumatic brain injury (TBI), and demyelination in Krabbe disease. We conclude by discussing the therapeutic potential of targeting the J2 prostaglandin pathway to prevent/delay neurodegeneration associated with neuroinflammation. In this context, we suggest a shift from the traditional view that cyclooxygenases are the most appropriate targets to treat neuroinflammation, to the notion that J2 prostaglandin pathways and other neurotoxic prostaglandins downstream from cyclooxygenases, would offer significant benefits as more effective therapeutic targets to treat chronic neurodegenerative diseases, while minimizing adverse side effects.
Keywords: J2 prostaglandins; UPP; mitochondria; neurodegeneration; neuroinflammation.
Figures






References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials