

Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome
- PMID: 25629079
- PMCID: PMC4299715
- DOI: 10.1002/mgg3.115
Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome
Abstract
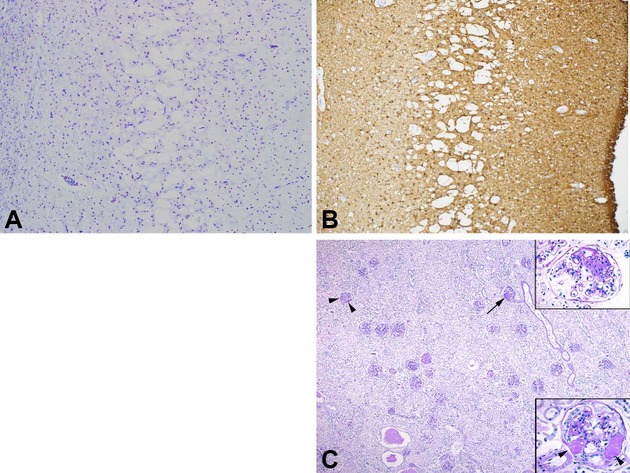
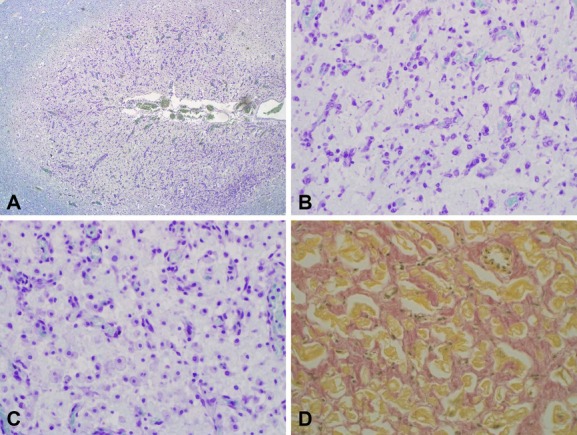
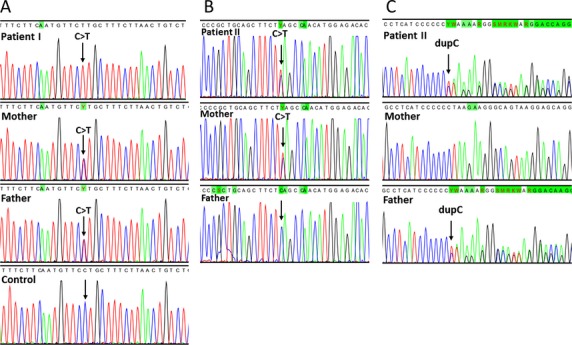
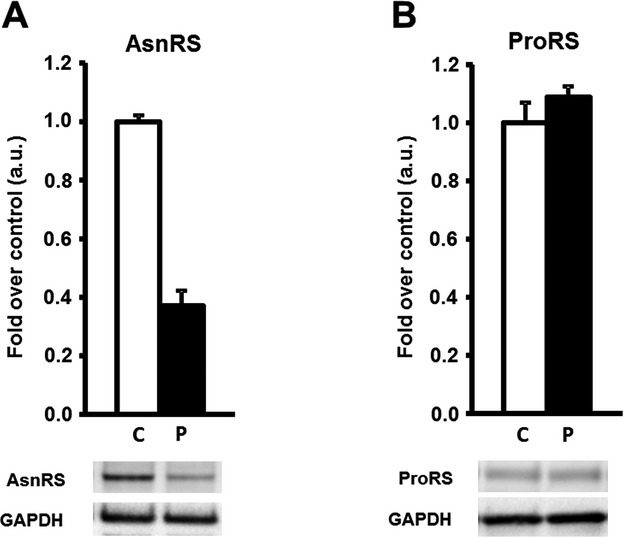
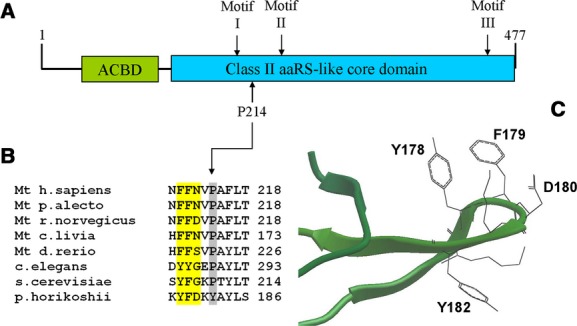
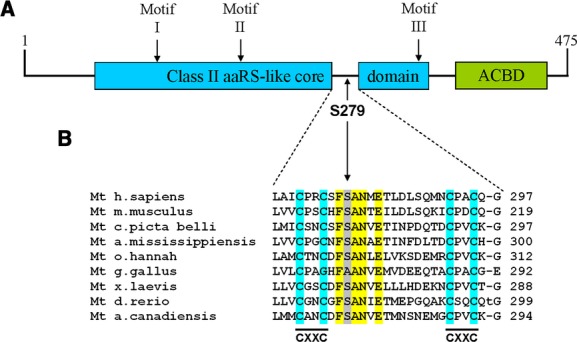
Alpers syndrome is a progressive neurodegenerative disorder that presents in infancy or early childhood and is characterized by diffuse degeneration of cerebral gray matter. While mutations in POLG1, the gene encoding the gamma subunit of the mitochondrial DNA polymerase, have been associated with Alpers syndrome with liver failure (Alpers-Huttenlocher syndrome), the genetic cause of Alpers syndrome in most patients remains unidentified. With whole exome sequencing we have identified mutations in NARS2 and PARS2, the genes encoding the mitochondrial asparaginyl-and prolyl-tRNA synthetases, in two patients with Alpers syndrome. One of the patients was homozygous for a missense mutation (c.641C>T, p.P214L) in NARS2. The affected residue is predicted to be located in the stem of a loop that participates in dimer interaction. The other patient was compound heterozygous for a one base insertion (c.1130dupC, p.K378 fs*1) that creates a premature stop codon and a missense mutation (c.836C>T, p.S279L) located in a conserved motif of unknown function in PARS2. This report links for the first time mutations in these genes to human disease in general and to Alpers syndrome in particular.
Keywords: Alpers syndrome; NARS2; PARS2; whole exome sequencing.
Figures
References
-
- Alpers BJ. Diffuse progressive degeneration of the gray matter of the cerebrum. Arch. Neurol. Psychiatry. 1931;25:469–505.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
