

A common gene signature across multiple studies relate biomarkers and functional regulation in tolerance to renal allograft
- PMID: 25629549
- PMCID: PMC4424816
- DOI: 10.1038/ki.2014.395
A common gene signature across multiple studies relate biomarkers and functional regulation in tolerance to renal allograft
Abstract
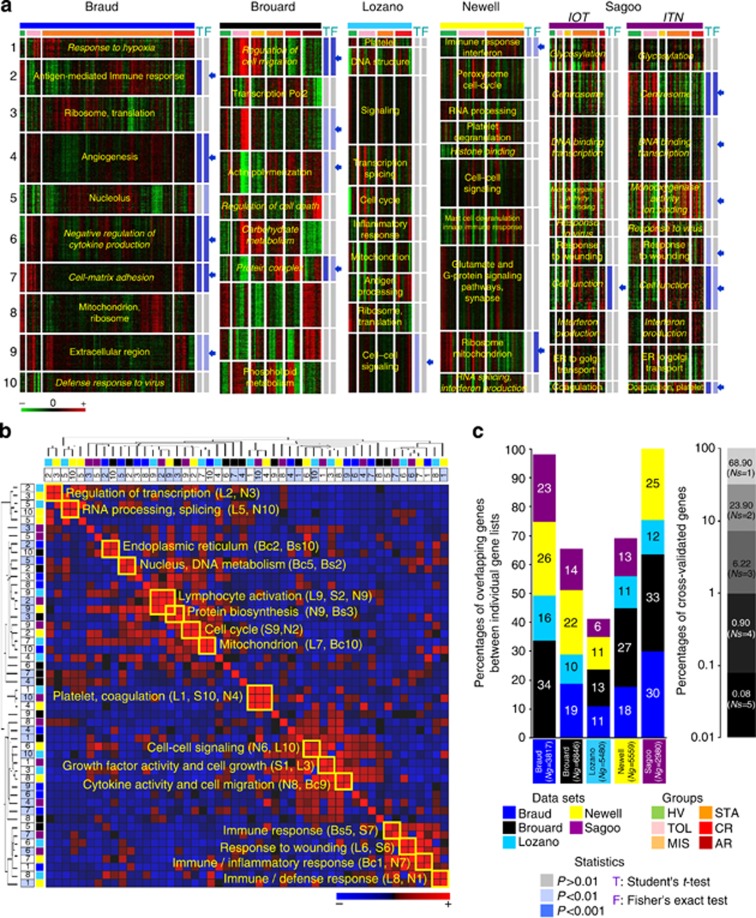
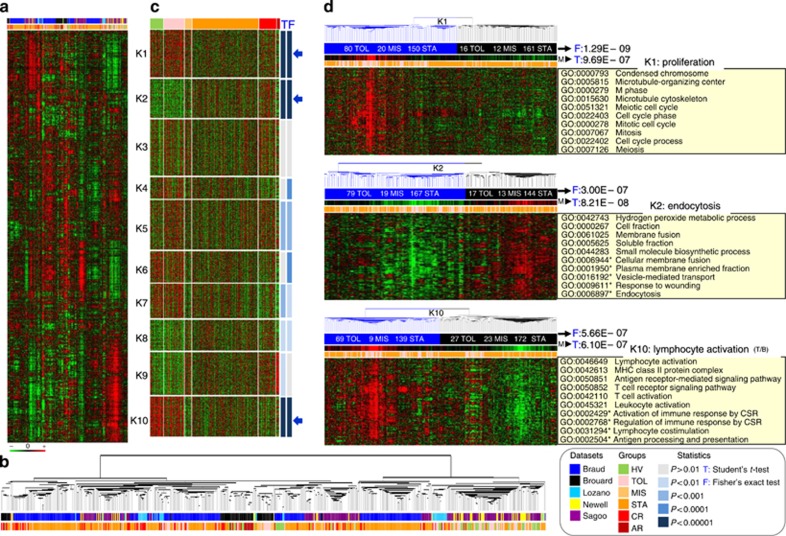
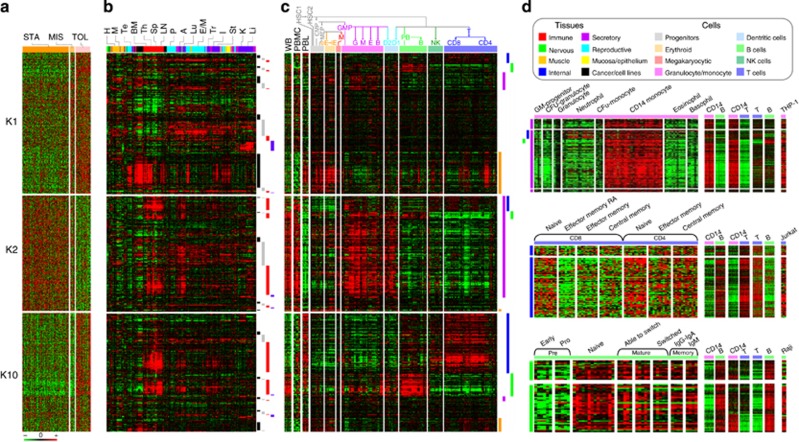
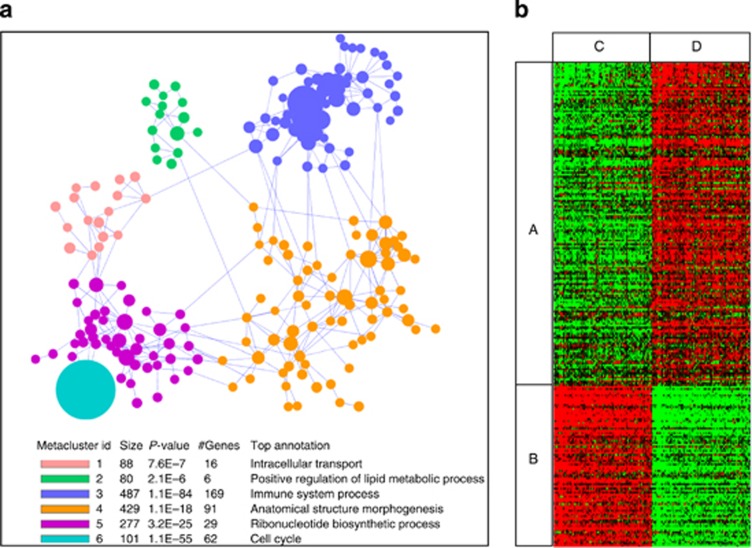
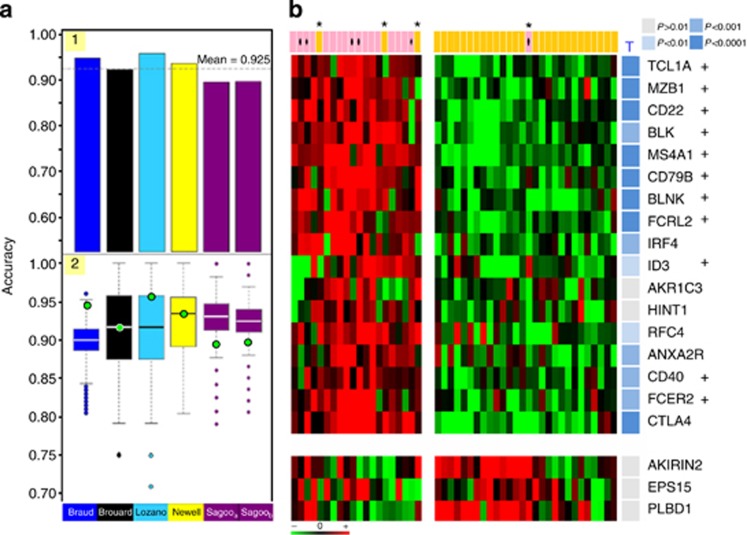
Patients tolerant to a kidney graft display a specific blood cell transcriptional pattern but results from five different studies were inconsistent, raising the question of relevance for future clinical application. To resolve this, we sought to identify a common gene signature, specific functional and cellular components, and discriminating biomarkers for tolerance following kidney transplantation. A meta-analysis of studies identified a robust gene signature involving proliferation of B and CD4 T cells, and inhibition of CD14 monocyte related functions among 96 tolerant samples. This signature was further supported through a cross-validation approach, yielding 92.5% accuracy independent of the study of origin. Experimental validation, performed on new tolerant samples and using a selection of the top-20 biomarkers, returned 91.7% of good classification. Beyond the confirmation of B-cell involvement, our data also indicated participation of other cell subsets in tolerance. Thus, the use of the top 20 biomarkers, mostly centered on B cells, may provide a common and standardized tool towards personalized medicine for the monitoring of tolerant or low-risk patients among kidney allotransplant recipients. These data point to a global preservation of genes favoring the maintenance of a homeostatic and 'healthy' environment in tolerant patients and may contribute to a better understanding of tolerance maintenance mechanisms.
Figures

Comment in
-
Telling the tiger by its stripes: mapping the genomics of kidney graft tolerance in real time.Kidney Int. 2015 May;87(5):875-7. doi: 10.1038/ki.2015.66. Kidney Int. 2015. PMID: 25951065
Similar articles
-
Gene expression signature of tolerance and lymphocyte subsets in stable renal transplants: results of a cross-sectional study.Transpl Immunol. 2014 Jun;31(1):11-6. doi: 10.1016/j.trim.2014.04.008. Epub 2014 May 5. Transpl Immunol. 2014. PMID: 24810779
-
Biomarkers of operational tolerance following kidney transplantation - The immune tolerance network studies of spontaneously tolerant kidney transplant recipients.Hum Immunol. 2018 May;79(5):380-387. doi: 10.1016/j.humimm.2018.02.007. Epub 2018 Feb 12. Hum Immunol. 2018. PMID: 29448053 Free PMC article. Review.
-
Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans.J Clin Invest. 2010 Jun;120(6):1848-61. doi: 10.1172/JCI39922. Epub 2010 May 24. J Clin Invest. 2010. PMID: 20501943 Free PMC article.
-
Telling the tiger by its stripes: mapping the genomics of kidney graft tolerance in real time.Kidney Int. 2015 May;87(5):875-7. doi: 10.1038/ki.2015.66. Kidney Int. 2015. PMID: 25951065
-
Urine biomarkers informative of human kidney allograft rejection and tolerance.Hum Immunol. 2018 May;79(5):343-355. doi: 10.1016/j.humimm.2018.01.006. Epub 2018 Jan 31. Hum Immunol. 2018. PMID: 29366869 Review.
Cited by
-
Harnessing Mechanisms of Immune Tolerance to Improve Outcomes in Solid Organ Transplantation: A Review.Front Immunol. 2021 Jun 10;12:688460. doi: 10.3389/fimmu.2021.688460. eCollection 2021. Front Immunol. 2021. PMID: 34177941 Free PMC article. Review.
-
Emergent Transcriptomic Technologies and Their Role in the Discovery of Biomarkers of Liver Transplant Tolerance.Front Immunol. 2015 Jun 22;6:304. doi: 10.3389/fimmu.2015.00304. eCollection 2015. Front Immunol. 2015. PMID: 26157438 Free PMC article. Review.
-
Immune monitoring as prerequisite for transplantation tolerance trials.Clin Exp Immunol. 2017 Aug;189(2):158-170. doi: 10.1111/cei.12988. Epub 2017 Jun 23. Clin Exp Immunol. 2017. PMID: 28518214 Free PMC article. Review.
-
An Unbiased Machine Learning Exploration Reveals Gene Sets Predictive of Allograft Tolerance After Kidney Transplantation.Front Immunol. 2021 Jul 8;12:695806. doi: 10.3389/fimmu.2021.695806. eCollection 2021. Front Immunol. 2021. PMID: 34305931 Free PMC article.
-
Identification of Candidate Biomarkers for Transplant Rejection from Transcriptome Data: A Systematic Review.Mol Diagn Ther. 2019 Aug;23(4):439-458. doi: 10.1007/s40291-019-00397-y. Mol Diagn Ther. 2019. PMID: 31054051
References
-
- Pascual M, Theruvath T, Kawai T, et al. Strategies to improve long-term outcomes after renal transplantation. N Engl J Med. 2002;346:580–590. - PubMed
-
- Dantal J, Hourmant M, Cantarovich D, et al. Effect of long-term immunosuppression in kidney-graft recipients on cancer incidence: randomised comparison of two cyclosporin regimens. Lancet. 1998;351:623–628. - PubMed
-
- Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357:2601–2614. - PubMed
-
- Soulillou JP, Giral M. Controlling the incidence of infection and malignancy by modifying immunosuppression. Transplantation. 2001;72:S89–S93. - PubMed
-
- Wimmer CD, Rentsch M, Crispin A, et al. The janus face of immunosuppression - de novo malignancy after renal transplantation: the experience of the Transplantation Center Munich. Kidney Int. 2007;71:1271–1278. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
