

Ten things you should know about protein kinases: IUPHAR Review 14
- PMID: 25630872
- PMCID: PMC4439867
- DOI: 10.1111/bph.13096
Ten things you should know about protein kinases: IUPHAR Review 14
Abstract
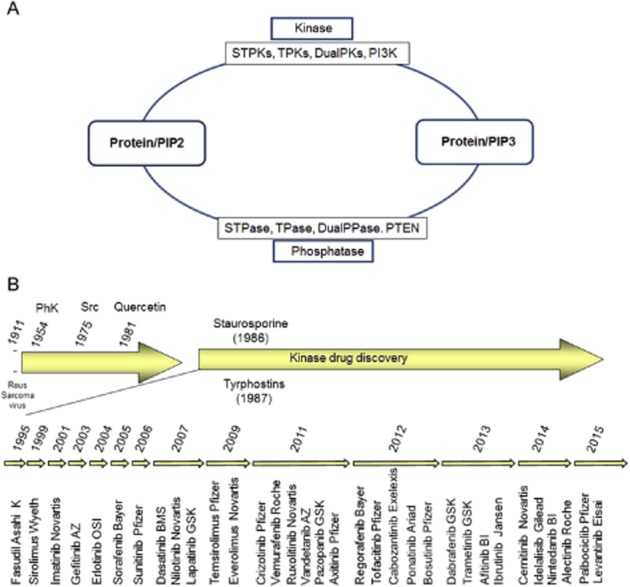
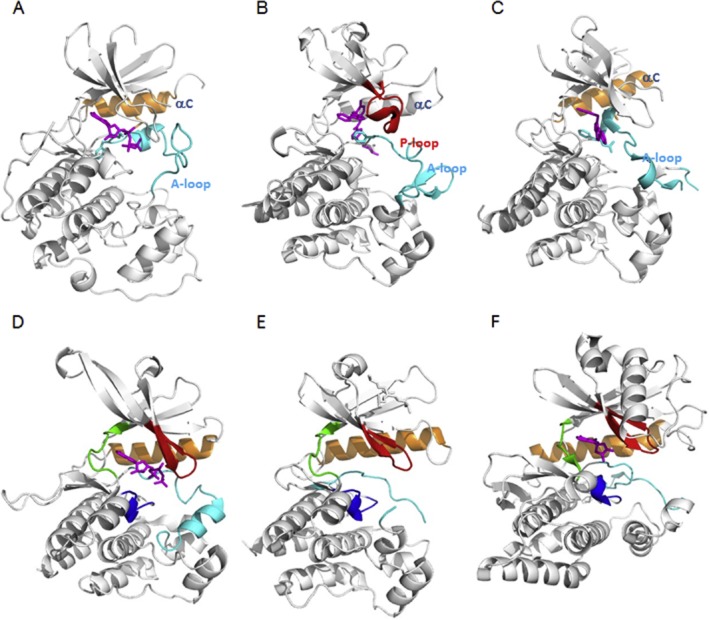
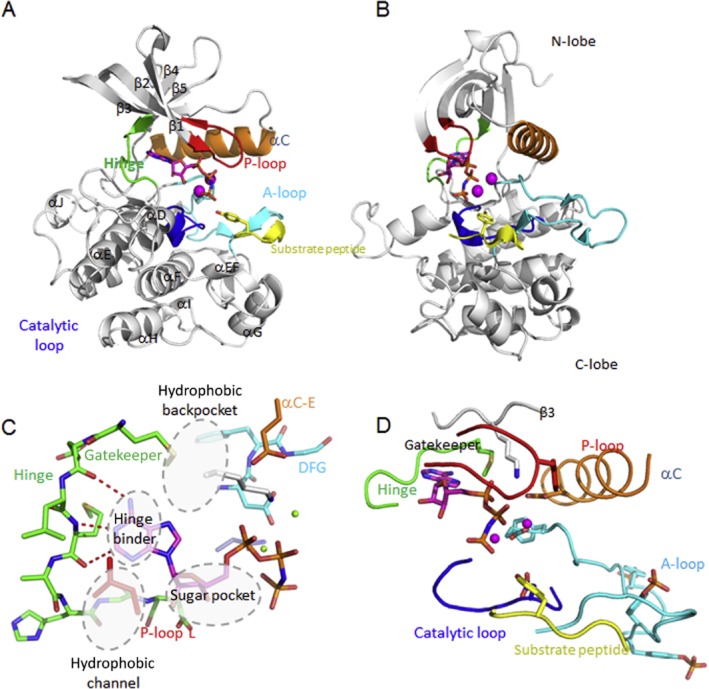
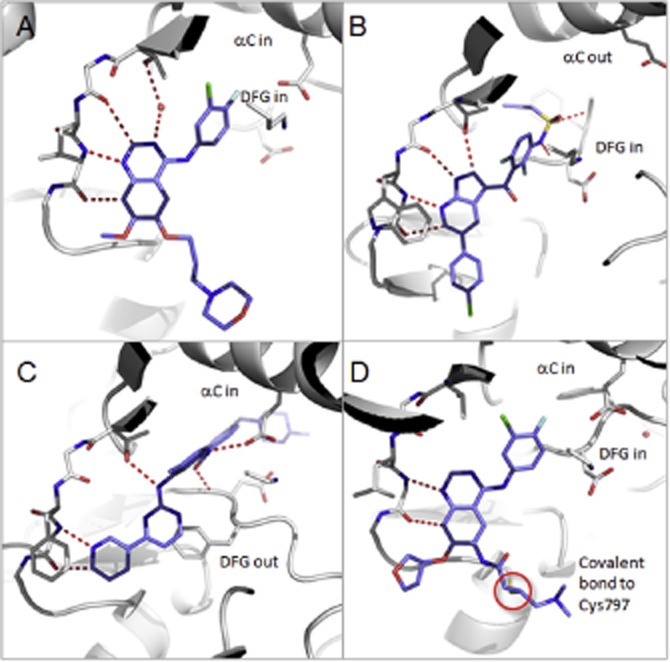
Many human malignancies are associated with aberrant regulation of protein or lipid kinases due to mutations, chromosomal rearrangements and/or gene amplification. Protein and lipid kinases represent an important target class for treating human disorders. This review focus on 'the 10 things you should know about protein kinases and their inhibitors', including a short introduction on the history of protein kinases and their inhibitors and ending with a perspective on kinase drug discovery. Although the '10 things' have been, to a certain extent, chosen arbitrarily, they cover in a comprehensive way the past and present efforts in kinase drug discovery and summarize the status quo of the current kinase inhibitors as well as knowledge about kinase structure and binding modes. Besides describing the potentials of protein kinase inhibitors as drugs, this review also focus on their limitations, particularly on how to circumvent emerging resistance against kinase inhibitors in oncological indications.
© 2015 The British Pharmacological Society.
Figures
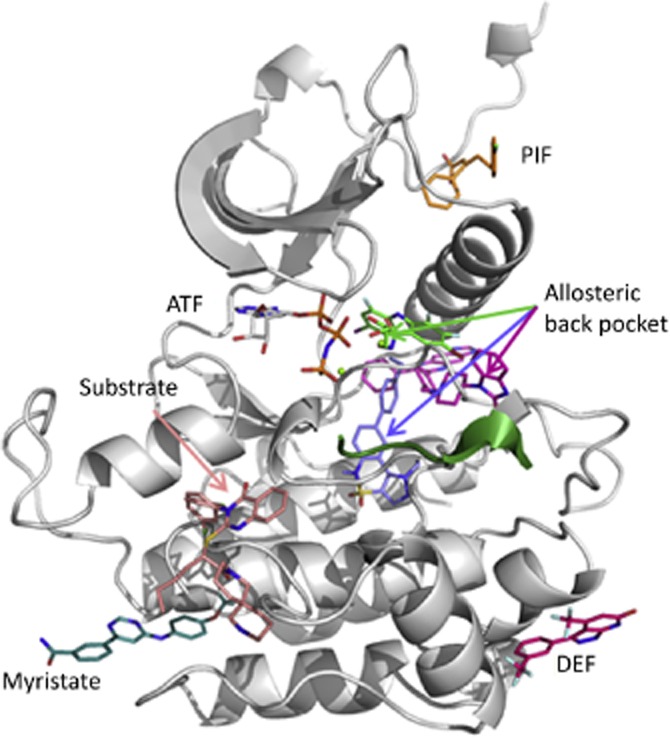
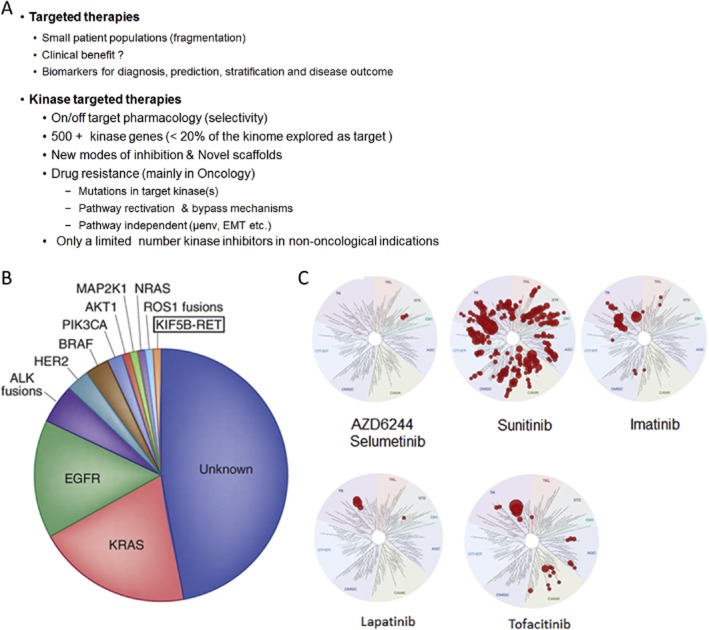
References
-
- Adrian FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95–102. - PubMed
-
- Akinleye A, Furqan M, Adekunle O. Ibrutinib and indolent B-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2014;14:253–260. - PubMed
-
- Albrecht BK, Harmange JC, Bauer D, Berry L, Bode C, Boezio AA, et al. Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J Med Chem. 2008;51:2879–2882. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
