

FXYD2, a γ subunit of Na⁺, K⁺-ATPase, maintains persistent mechanical allodynia induced by inflammation
- PMID: 25633594
- PMCID: PMC4349241
- DOI: 10.1038/cr.2015.12
FXYD2, a γ subunit of Na⁺, K⁺-ATPase, maintains persistent mechanical allodynia induced by inflammation
Abstract
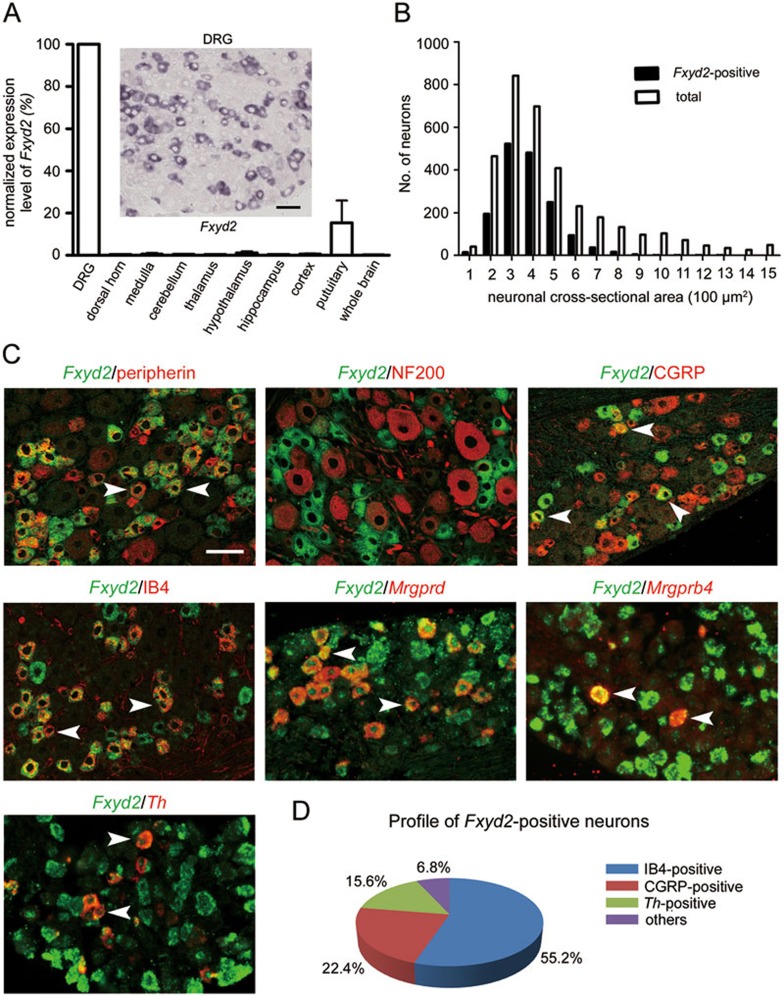
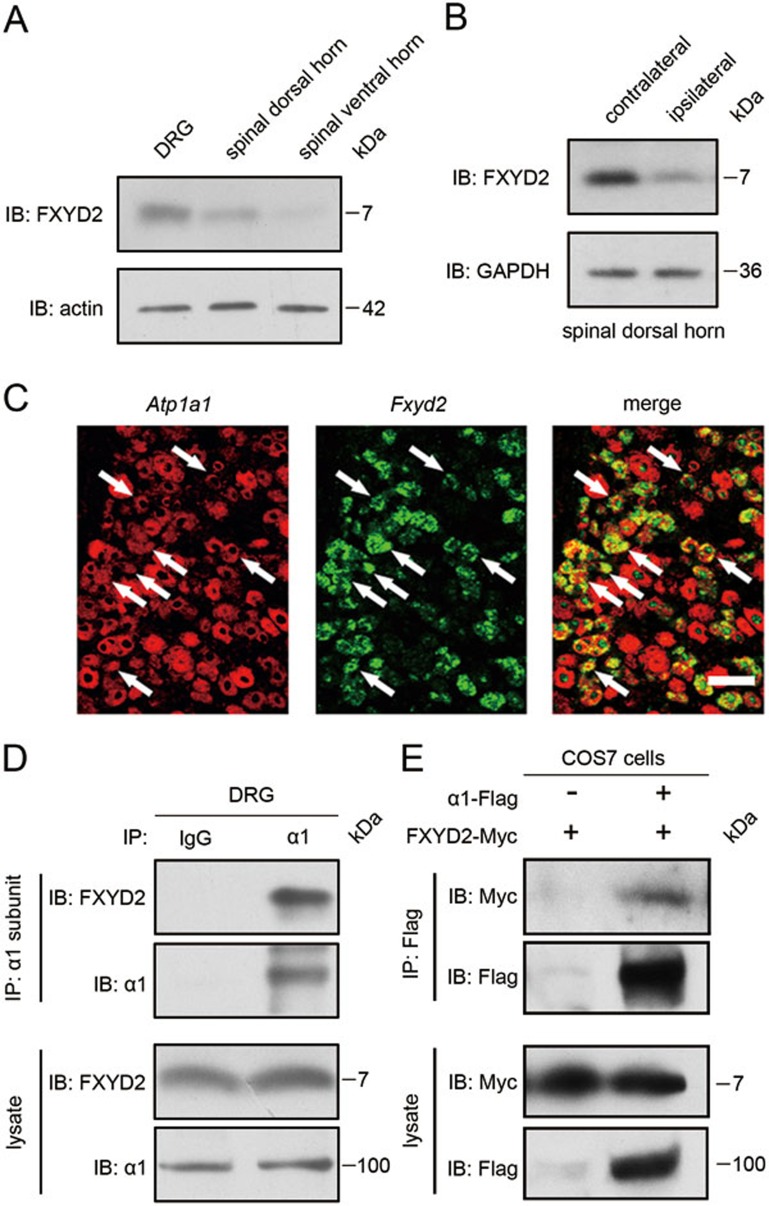
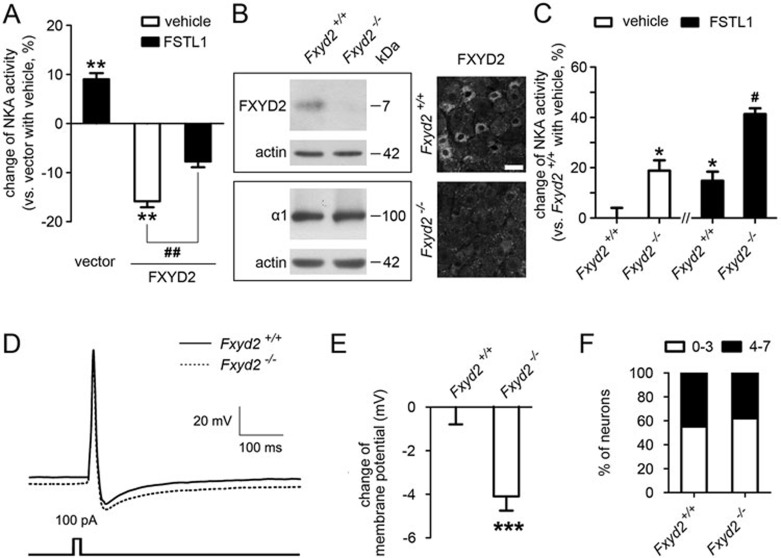
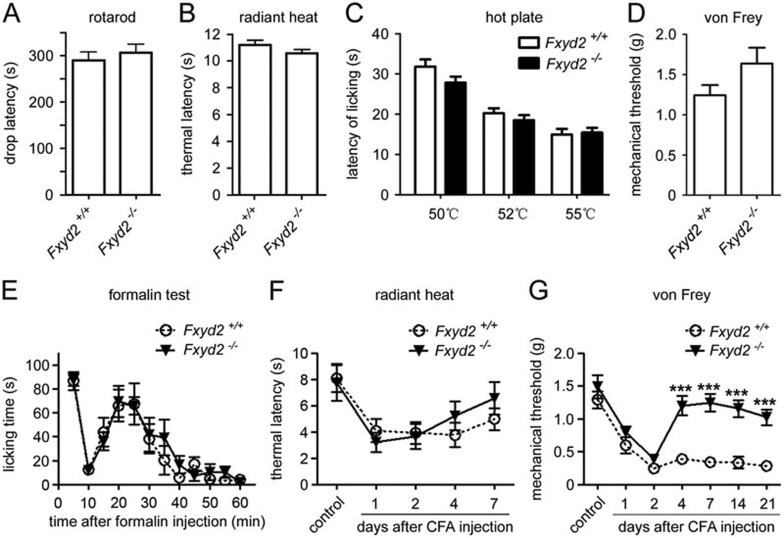
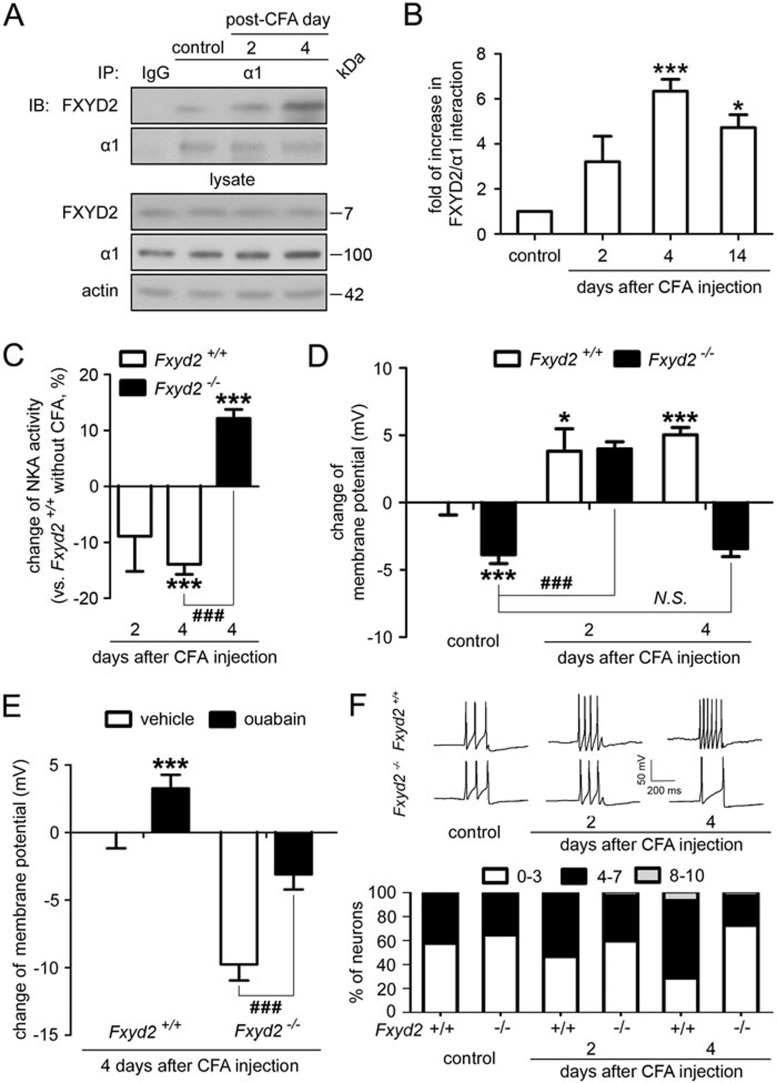
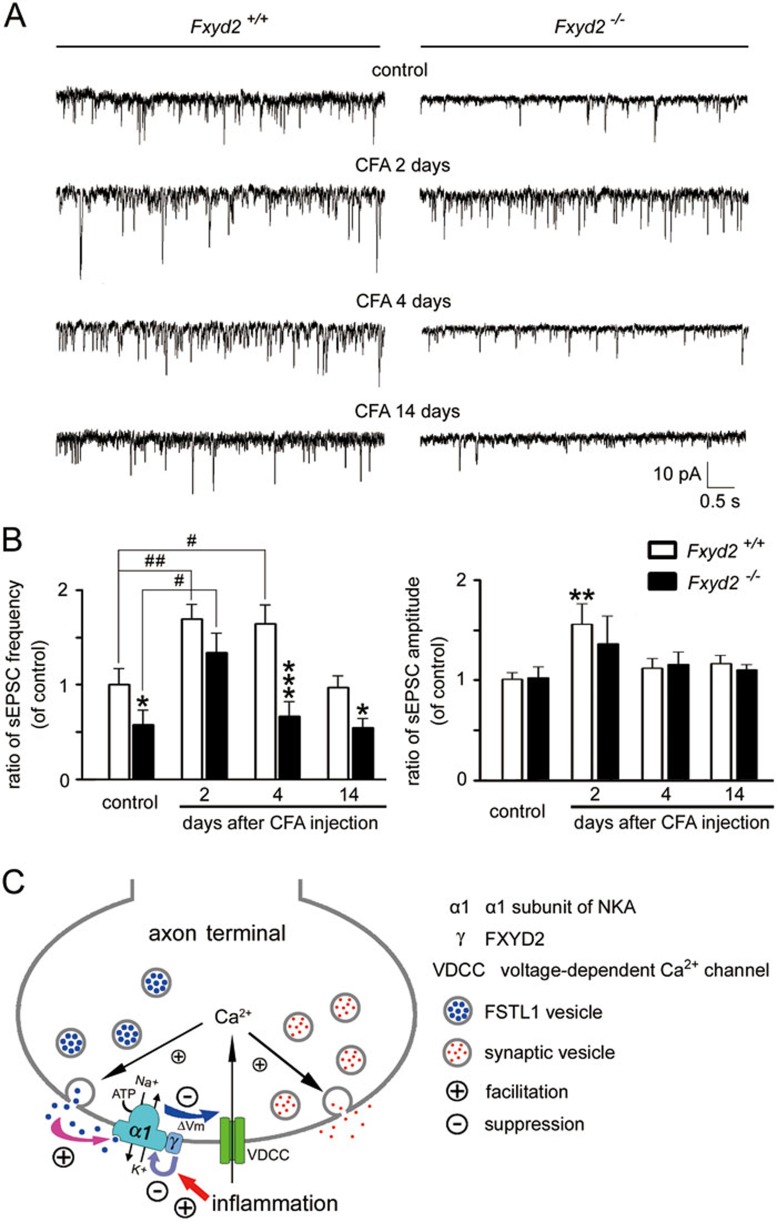
Na⁺, K⁺-ATPase (NKA) is required to generate the resting membrane potential in neurons. Nociceptive afferent neurons express not only the α and β subunits of NKA but also the γ subunit FXYD2. However, the neural function of FXYD2 is unknown. The present study shows that FXYD2 in nociceptive neurons is necessary for maintaining the mechanical allodynia induced by peripheral inflammation. FXYD2 interacted with α1NKA and negatively regulated the NKA activity, depolarizing the membrane potential of nociceptive neurons. Mechanical allodynia initiated in FXYD2-deficient mice was abolished 4 days after inflammation, whereas it persisted for at least 3 weeks in wild-type mice. Importantly, the FXYD2/α1NKA interaction gradually increased after inflammation and peaked on day 4 post inflammation, resulting in reduction of NKA activity, depolarization of neuron membrane and facilitation of excitatory afferent neurotransmission. Thus, the increased FXYD2 activity may be a fundamental mechanism underlying the persistent hypersensitivity to pain induced by inflammation.
Figures
Similar articles
-
Regulation of the Na,K-ATPase gamma-subunit FXYD2 by Runx1 and Ret signaling in normal and injured non-peptidergic nociceptive sensory neurons.PLoS One. 2012;7(1):e29852. doi: 10.1371/journal.pone.0029852. Epub 2012 Jan 13. PLoS One. 2012. PMID: 22253804 Free PMC article.
-
Follistatin-like 1 suppresses sensory afferent transmission by activating Na+,K+-ATPase.Neuron. 2011 Mar 10;69(5):974-87. doi: 10.1016/j.neuron.2011.01.022. Neuron. 2011. PMID: 21382556
-
Glutamate transporter GLAST/EAAT1 directs cell surface expression of FXYD2/gamma subunit of Na, K-ATPase in human fetal astrocytes.Neurochem Int. 2007 Jun;50(7-8):916-20. doi: 10.1016/j.neuint.2006.12.015. Epub 2007 Jan 14. Neurochem Int. 2007. PMID: 17316900
-
Renal Mg handling, FXYD2 and the central role of the Na,K-ATPase.Physiol Rep. 2018 Sep;6(17):e13843. doi: 10.14814/phy2.13843. Physiol Rep. 2018. PMID: 30175537 Free PMC article. Review.
-
Beneficial Renal and Pancreatic Phenotypes in a Mouse Deficient in FXYD2 Regulatory Subunit of Na,K-ATPase.Front Physiol. 2016 Mar 7;7:88. doi: 10.3389/fphys.2016.00088. eCollection 2016. Front Physiol. 2016. PMID: 27014088 Free PMC article. Review.
Cited by
-
Application of Proteomics and Metabonomics to Reveal the Molecular Basis of Atractylodis Macrocephalae Rhizome for Ameliorating Hypothyroidism Instead of Hyperthyroidism.Front Pharmacol. 2021 Apr 20;12:664319. doi: 10.3389/fphar.2021.664319. eCollection 2021. Front Pharmacol. 2021. PMID: 33959028 Free PMC article.
-
Deletion of the Gene Encoding the NMDA Receptor GluN1 Subunit in Schwann Cells Causes Ultrastructural Changes in Remak Bundles and Hypersensitivity in Pain Processing.J Neurosci. 2020 Nov 18;40(47):9121-9136. doi: 10.1523/JNEUROSCI.0663-20.2020. Epub 2020 Oct 13. J Neurosci. 2020. PMID: 33051351 Free PMC article.
-
Roads Less Traveled: Sexual Dimorphism and Mast Cell Contributions to Migraine Pathology.Front Immunol. 2016 Apr 19;7:140. doi: 10.3389/fimmu.2016.00140. eCollection 2016. Front Immunol. 2016. PMID: 27148260 Free PMC article. Review.
-
Low Dopamine D2 Receptor Expression Drives Gene Networks Related to GABA, cAMP, Growth and Neuroinflammation in Striatal Indirect Pathway Neurons.Biol Psychiatry Glob Open Sci. 2022 Sep 8;3(4):1104-1115. doi: 10.1016/j.bpsgos.2022.08.010. eCollection 2023 Oct. Biol Psychiatry Glob Open Sci. 2022. PMID: 37881572 Free PMC article.
-
Fxyd2 regulates Aδ- and C-fiber mechanosensitivity and is required for the maintenance of neuropathic pain.Sci Rep. 2016 Nov 2;6:36407. doi: 10.1038/srep36407. Sci Rep. 2016. PMID: 27805035 Free PMC article.
References
-
- Geering K. The functional role of the β-subunit in the maturation and intracellular transport of Na,K-ATPase. FEBS Lett. 1991;285:189–193. - PubMed
-
- Noguchi S, Mishina M, Kawamura M, Numa S. Expression of functional (Na+ + K+)-ATPase from cloned cDNAs. FEBS Lett. 1987;225:27–32. - PubMed
-
- Jewell EA, Lingrel JB. Comparison of the substrate dependence properties of the rat Na,K-ATPase 1, 2, and 3 isoforms expressed in HeLa cells. J Biol Chem. 1991;266:16925–16930. - PubMed
-
- Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–566. - PubMed
-
- Charnock JS, Post RL. Evidence of the mechanism of ouabain inhibition of cation-activated adenosine triphosphate. Nature. 1963;199:910–911. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
