

Highlighting nonlinear patterns in population genetics datasets
- PMID: 25633916
- PMCID: PMC4311249
- DOI: 10.1038/srep08140
Highlighting nonlinear patterns in population genetics datasets
Abstract
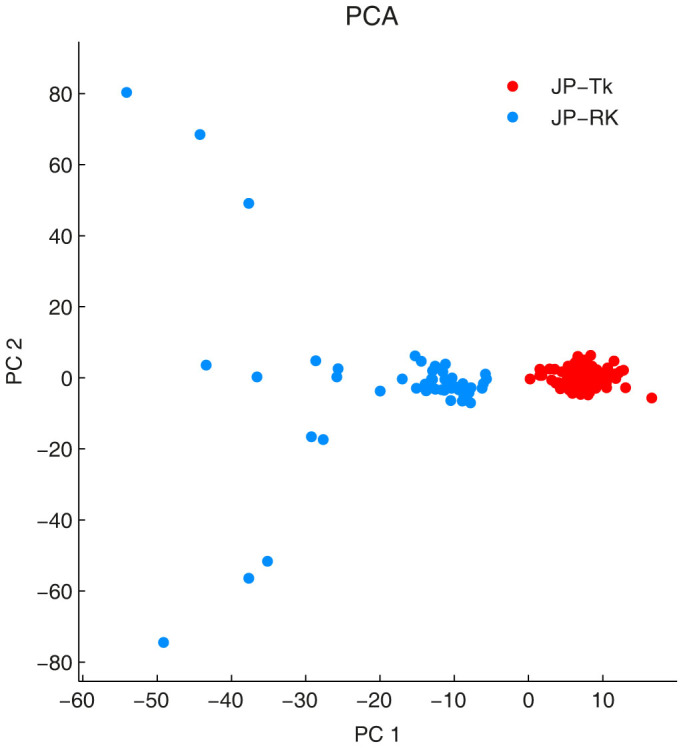
Detecting structure in population genetics and case-control studies is important, as it exposes phenomena such as ecoclines, admixture and stratification. Principal Component Analysis (PCA) is a linear dimension-reduction technique commonly used for this purpose, but it struggles to reveal complex, nonlinear data patterns. In this paper we introduce non-centred Minimum Curvilinear Embedding (ncMCE), a nonlinear method to overcome this problem. Our analyses show that ncMCE can separate individuals into ethnic groups in cases in which PCA fails to reveal any clear structure. This increased discrimination power arises from ncMCE's ability to better capture the phylogenetic signal in the samples, whereas PCA better reflects their geographic relation. We also demonstrate how ncMCE can discover interesting patterns, even when the data has been poorly pre-processed. The juxtaposition of PCA and ncMCE visualisations provides a new standard of analysis with utility for discovering and validating significant linear/nonlinear complementary patterns in genetic data.
Figures





References
-
- Jolliffe I. T. Principal Component Analysis. 489 (Springer, 2002).
-
- Menozzi P., Piazza A. & Cavalli-Sforza L. Synthetic maps of human gene frequencies in Europeans. Science. 201, 786–792 (1978). - PubMed
-
- Price A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
