

Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses
- PMID: 25633976
- PMCID: PMC4384744
- DOI: 10.7554/eLife.05378
Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses
Abstract
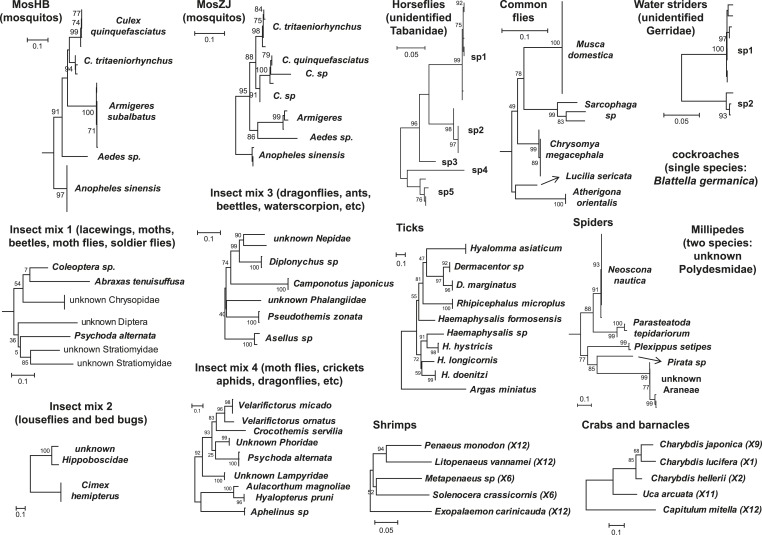
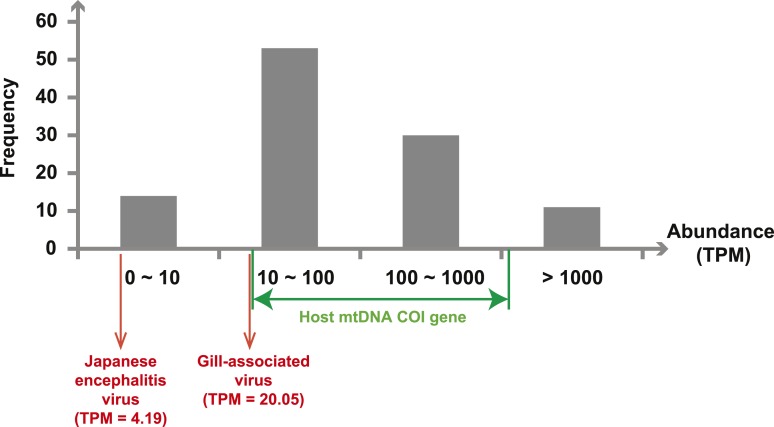
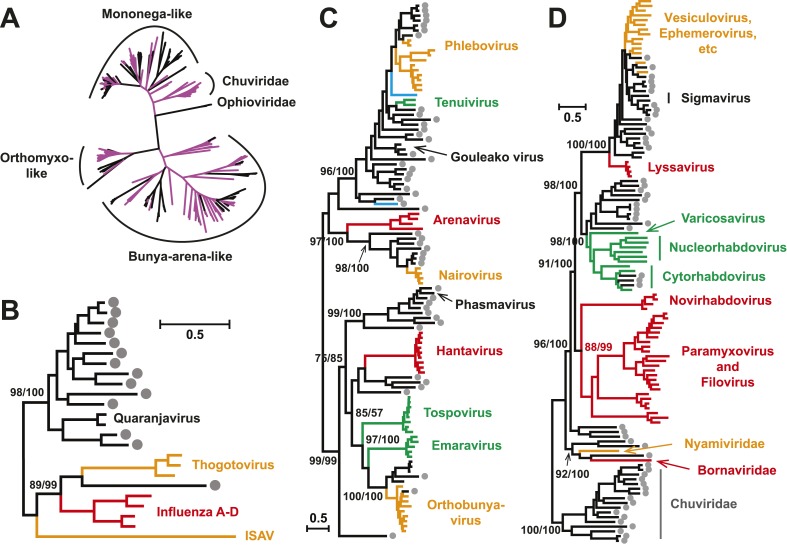
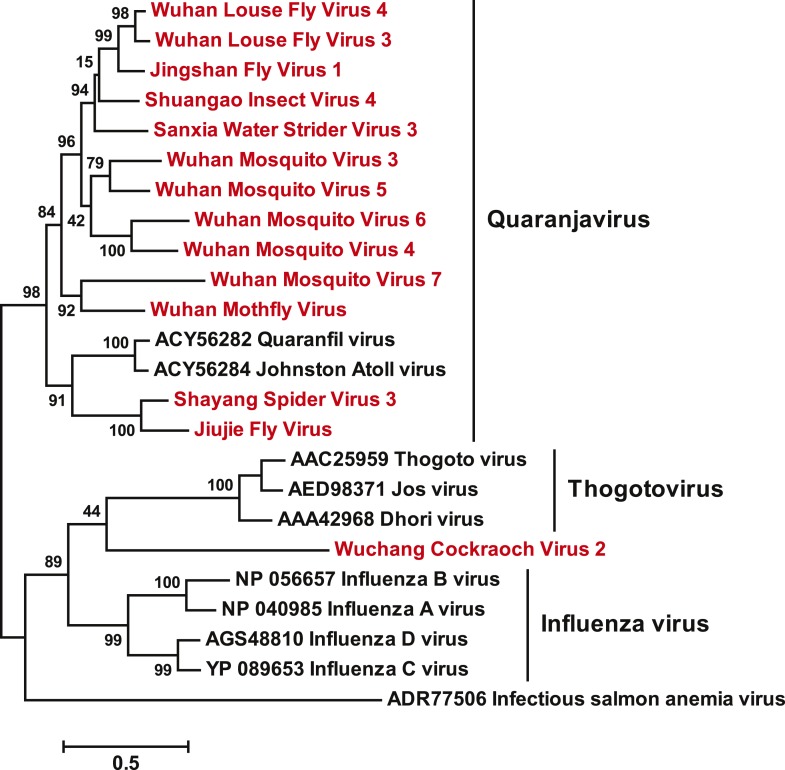
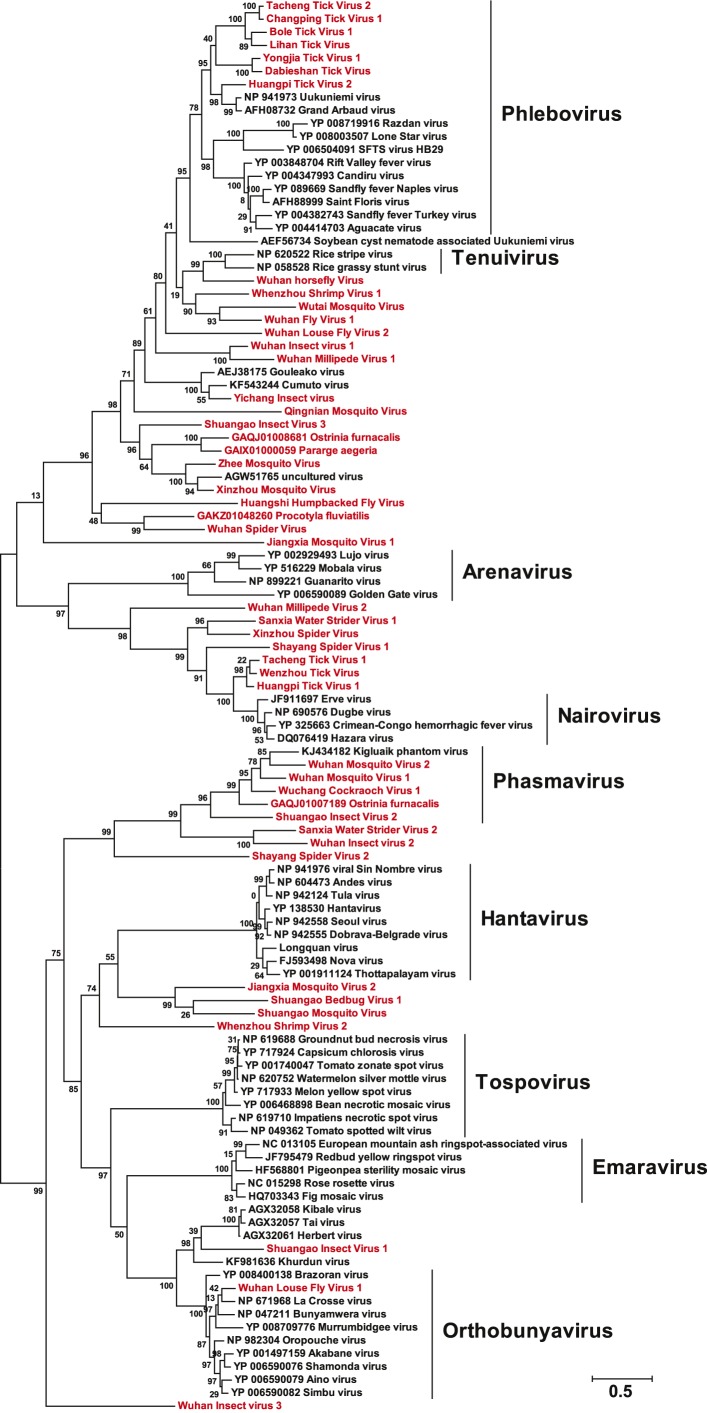
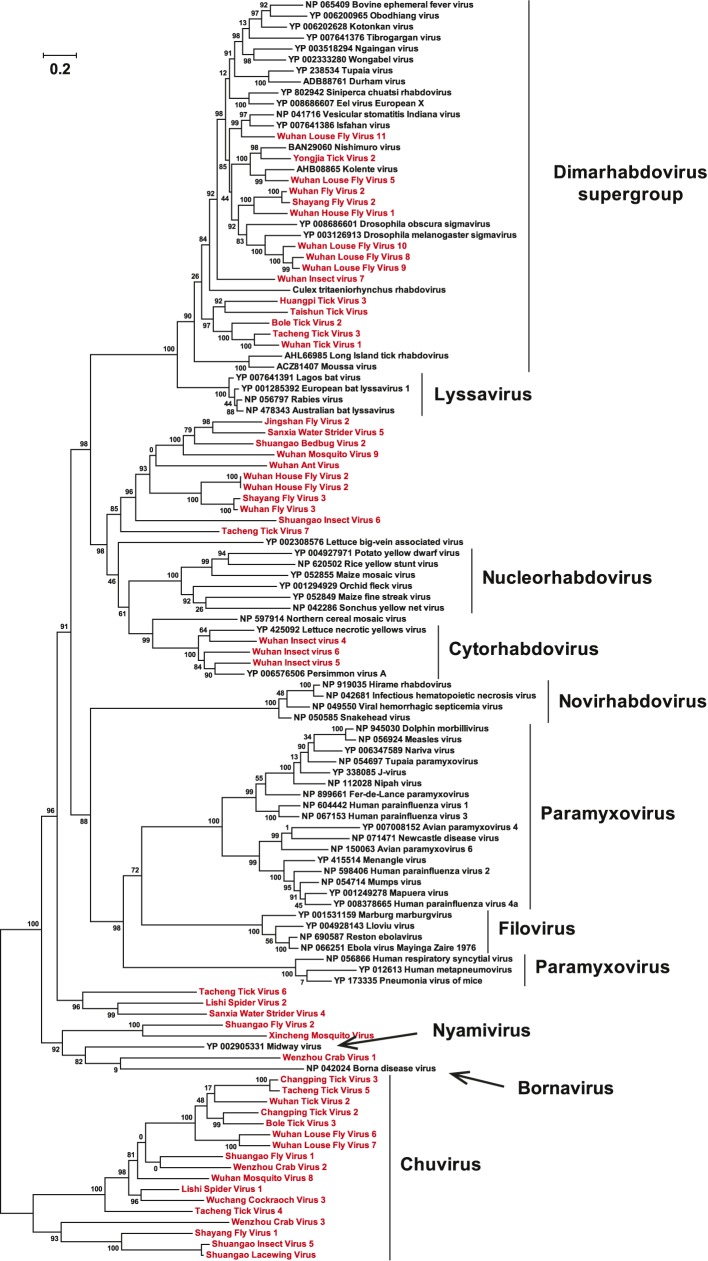
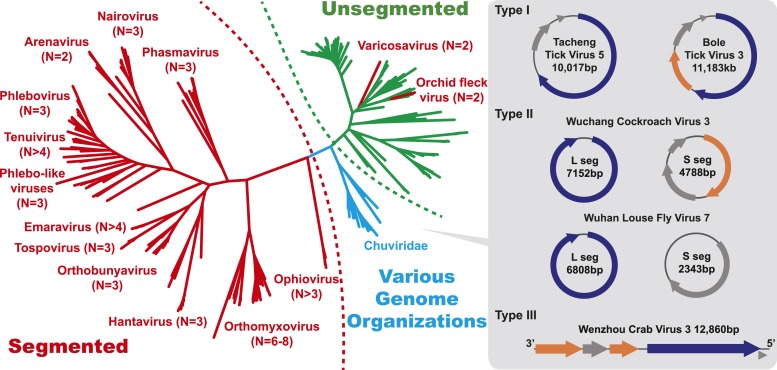
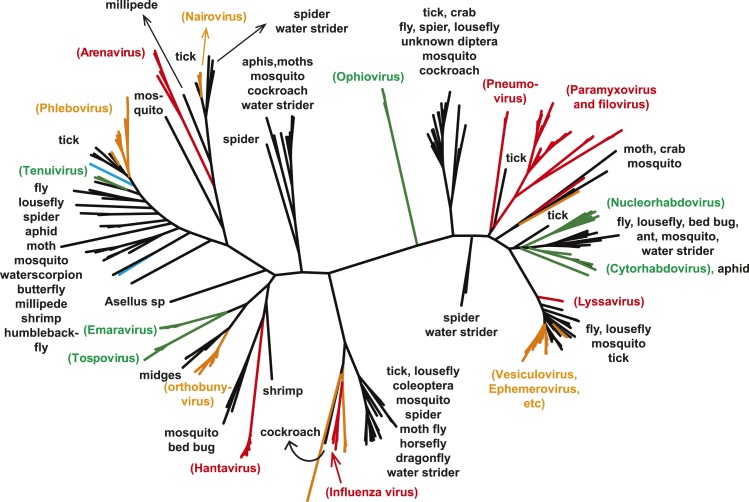
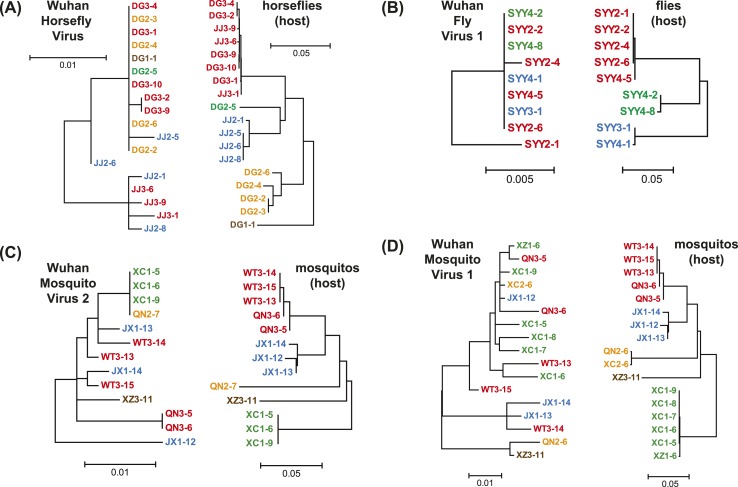
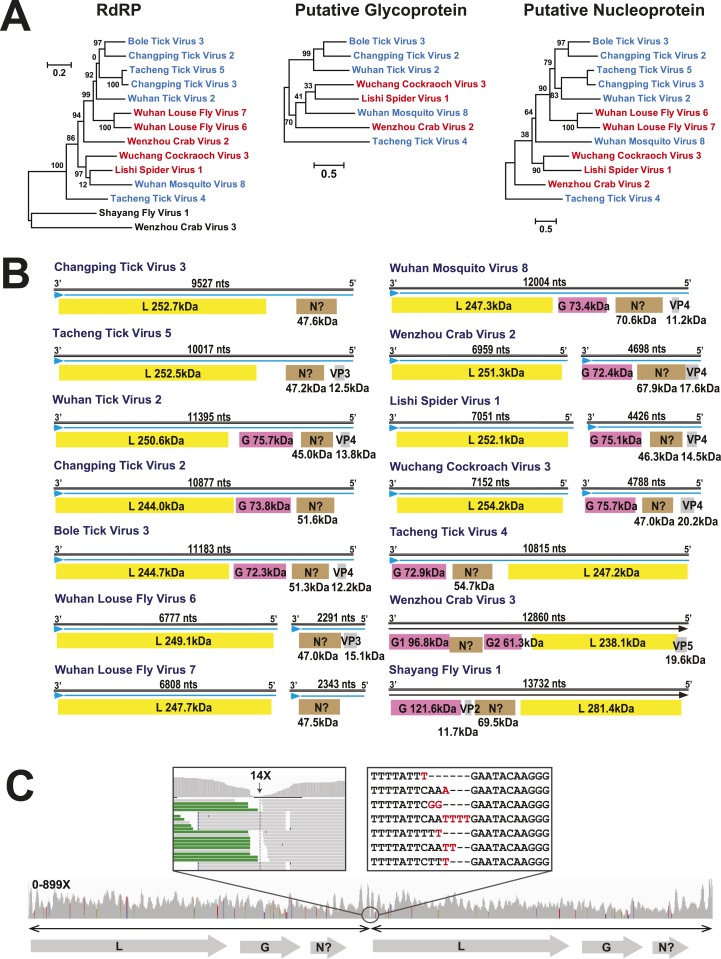
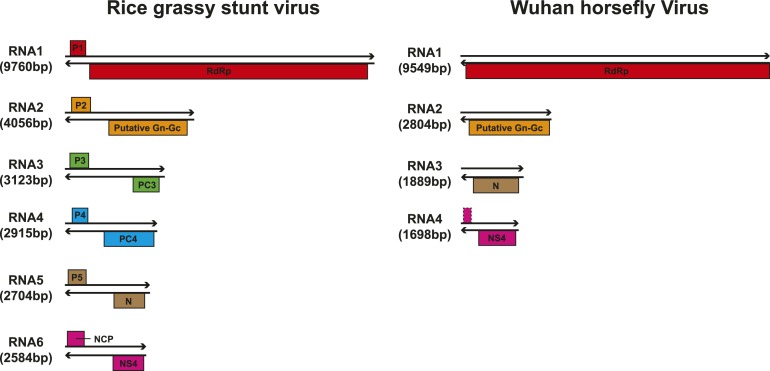
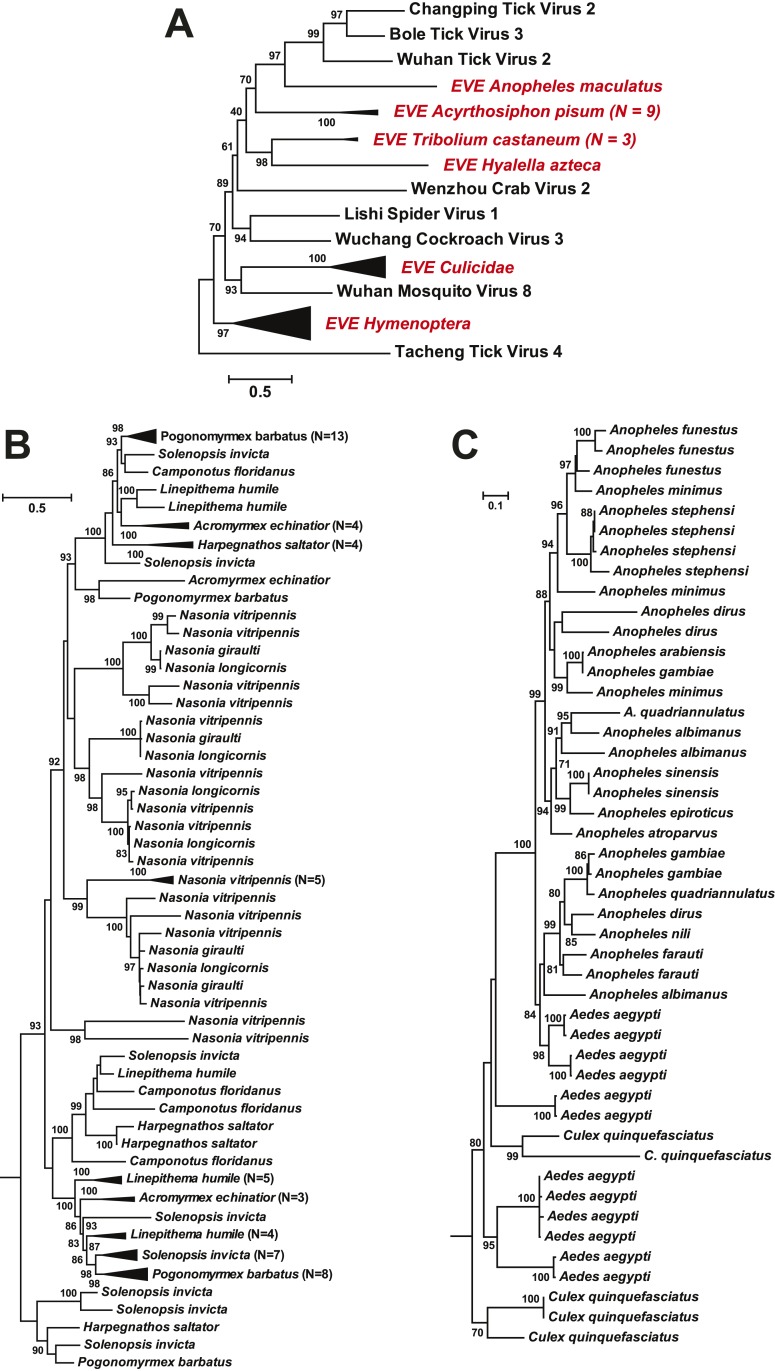
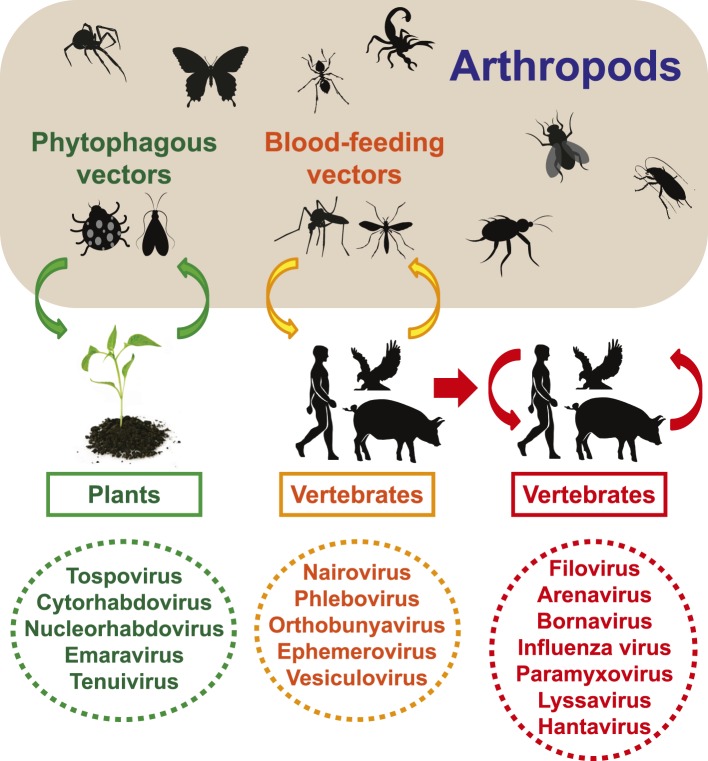
Although arthropods are important viral vectors, the biodiversity of arthropod viruses, as well as the role that arthropods have played in viral origins and evolution, is unclear. Through RNA sequencing of 70 arthropod species we discovered 112 novel viruses that appear to be ancestral to much of the documented genetic diversity of negative-sense RNA viruses, a number of which are also present as endogenous genomic copies. With this greatly enriched diversity we revealed that arthropods contain viruses that fall basal to major virus groups, including the vertebrate-specific arenaviruses, filoviruses, hantaviruses, influenza viruses, lyssaviruses, and paramyxoviruses. We similarly documented a remarkable diversity of genome structures in arthropod viruses, including a putative circular form, that sheds new light on the evolution of genome organization. Hence, arthropods are a major reservoir of viral genetic diversity and have likely been central to viral evolution.
Keywords: RNA virus; arthropods; evolution; infectious disease; microbiology; negative-sense; phylogeny; segmentation; viruses.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures


References
-
- Cook S, Chung BY, Bass D, Moureau G, Tang S, McAlister E, Culverwell CL, Glucksman E, Wang H, Brown TD, Gould EA, Harbach RE, de Lamballerie X, Firth AE. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLOS ONE. 2013;8:e80720. doi: 10.1371/journal.pone.0080720. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
