

Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort
- PMID: 25635037
- PMCID: PMC4386250
- DOI: 10.2215/CJN.06260614
Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort
Abstract
Background and objectives: Steroid-resistant nephrotic syndrome is a rare kidney disease involving either immune-mediated or genetic alterations of podocyte structure and function. The rare nature, heterogeneity, and slow evolution of the disorder are major obstacles to systematic genotype-phenotype, intervention, and outcome studies, hampering the development of evidence-based diagnostic and therapeutic concepts. To overcome these limitations, the PodoNet Consortium has created an international registry for congenital nephrotic syndrome and childhood-onset steroid-resistant nephrotic syndrome.
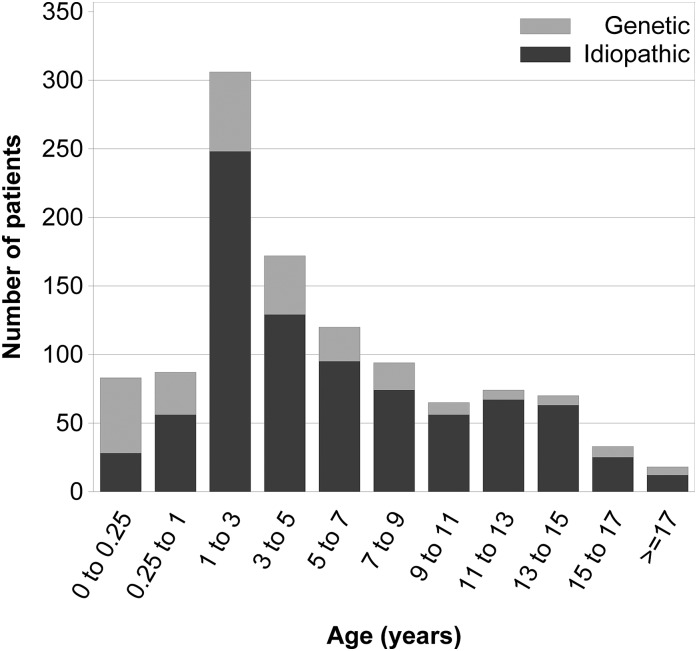
Design, setting, participants, & measurements: Since August of 2009, clinical, biochemical, genetic, and histopathologic information was collected both retrospectively and prospectively from 1655 patients with childhood-onset steroid-resistant nephrotic syndrome, congenital nephrotic syndrome, or persistent subnephrotic proteinuria of likely genetic origin at 67 centers in 21 countries through an online portal.
Results: Steroid-resistant nephrotic syndrome manifested in the first 5 years of life in 64% of the patients. Congenital nephrotic syndrome accounted for 6% of all patients. Extrarenal abnormalities were reported in 17% of patients. The most common histopathologic diagnoses were FSGS (56%), minimal change nephropathy (21%), and mesangioproliferative GN (12%). Mutation screening was performed in 1174 patients, and a genetic disease cause was identified in 23.6% of the screened patients. Among 14 genes with reported mutations, abnormalities in NPHS2 (n=138), WT1 (n=48), and NPHS1 (n=41) were most commonly identified. The proportion of patients with a genetic disease cause decreased with increasing manifestation age: from 66% in congenital nephrotic syndrome to 15%-16% in schoolchildren and adolescents. Among various intensified immunosuppressive therapy protocols, calcineurin inhibitors and rituximab yielded consistently high response rates, with 40%-45% of patients achieving complete remission. Confirmation of a genetic diagnosis but not the histopathologic disease type was strongly predictive of intensified immunosuppressive therapy responsiveness. Post-transplant disease recurrence was noted in 25.8% of patients without compared with 4.5% (n=4) of patients with a genetic diagnosis.
Conclusions: The PodoNet cohort may serve as a source of reference for future clinical and genetic research in this rare but significant kidney disease.
Keywords: WT1; children; nephrin; podocin; podocytopathies.
Copyright © 2015 by the American Society of Nephrology.
Figures
References
-
- McKinney PA, Feltbower RG, Brocklebank JT, Fitzpatrick MM: Time trends and ethnic patterns of childhood nephrotic syndrome in Yorkshire, UK. Pediatr Nephrol 16: 1040–1044, 2001 - PubMed
-
- Kim JS, Bellew CA, Silverstein DM, Aviles DH, Boineau FG, Vehaskari VM: High incidence of initial and late steroid resistance in childhood nephrotic syndrome. Kidney Int 68: 1275–1281, 2005 - PubMed
-
- Mekahli D, Liutkus A, Ranchin B, Yu A, Bessenay L, Girardin E, Van Damme-Lombaerts R, Palcoux JB, Cachat F, Lavocat MP, Bourdat-Michel G, Nobili F, Cochat P: Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: A multicenter study. Pediatr Nephrol 24: 1525–1532, 2009 - PubMed
-
- Zagury A, Oliveira AL, Montalvão JA, Novaes RH, Sá VM, Moraes CA, Tavares MS: Steroid-resistant idiopathic nephrotic syndrome in children: Long-term follow-up and risk factors for end-stage renal disease. J Bras Neurol 35: 191–199, 2013 - PubMed
-
- Saleem MA: New developments in steroid-resistant nephrotic syndrome. Pediatr Nephrol 28: 699–709, 2013 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
