

Enhancer evolution across 20 mammalian species
- PMID: 25635462
- PMCID: PMC4313353
- DOI: 10.1016/j.cell.2015.01.006
Enhancer evolution across 20 mammalian species
Abstract
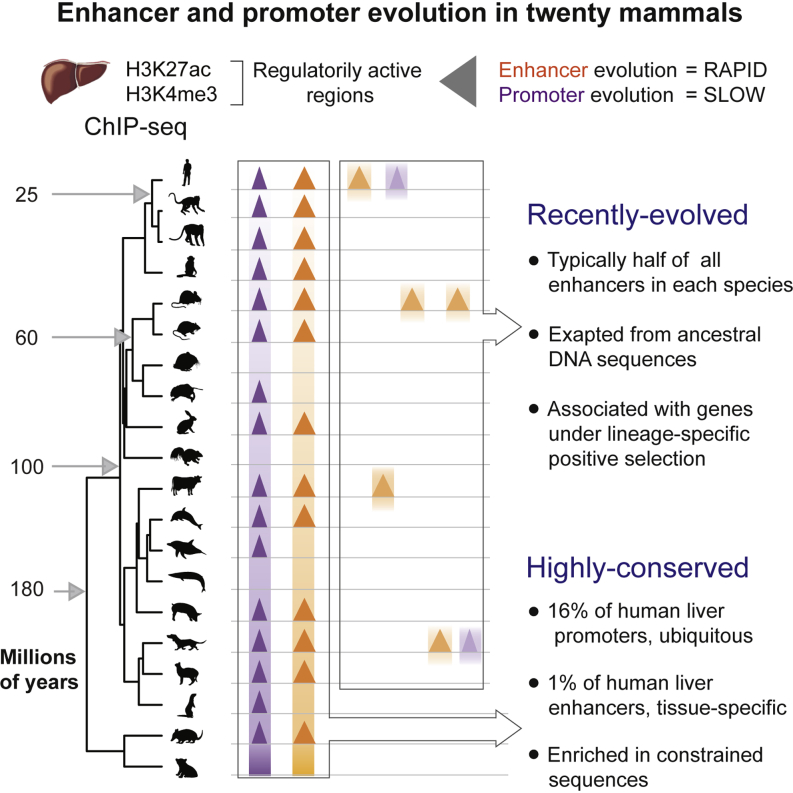
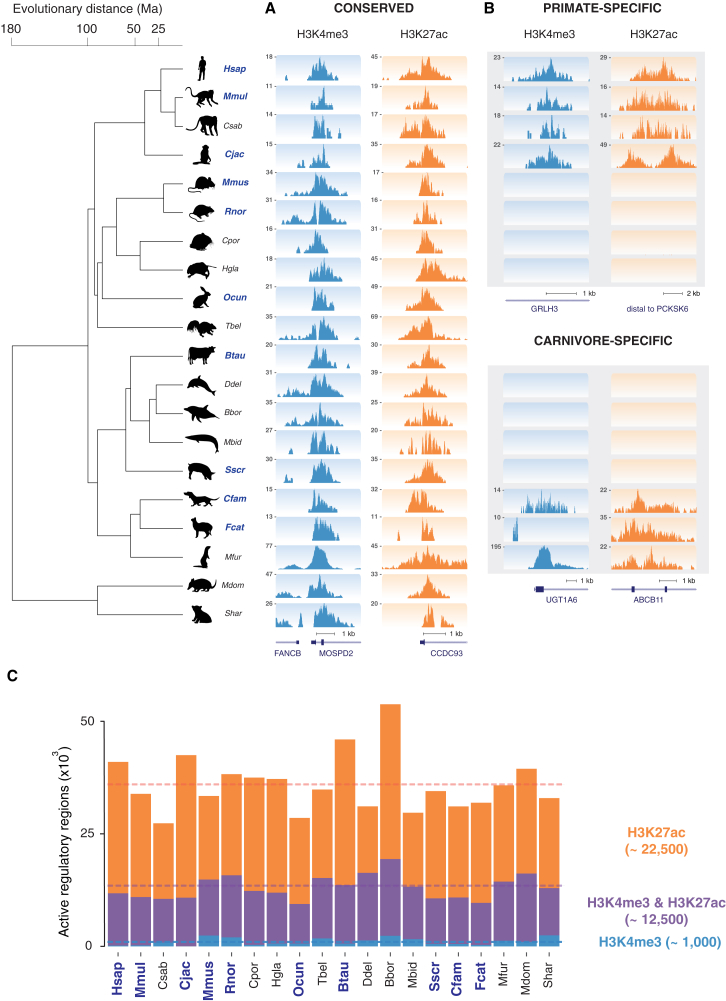
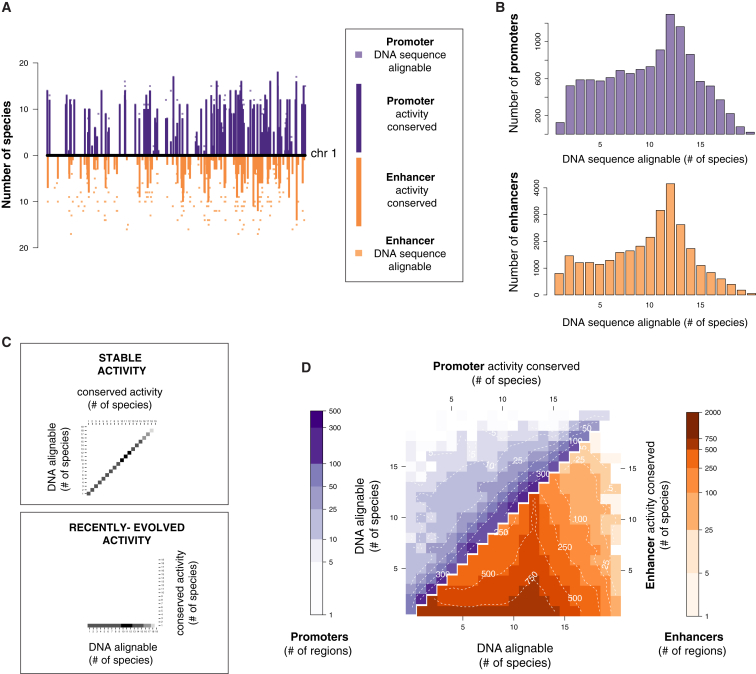
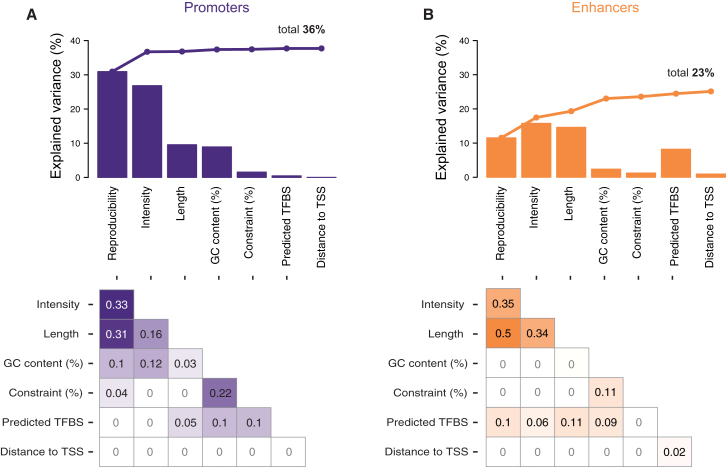
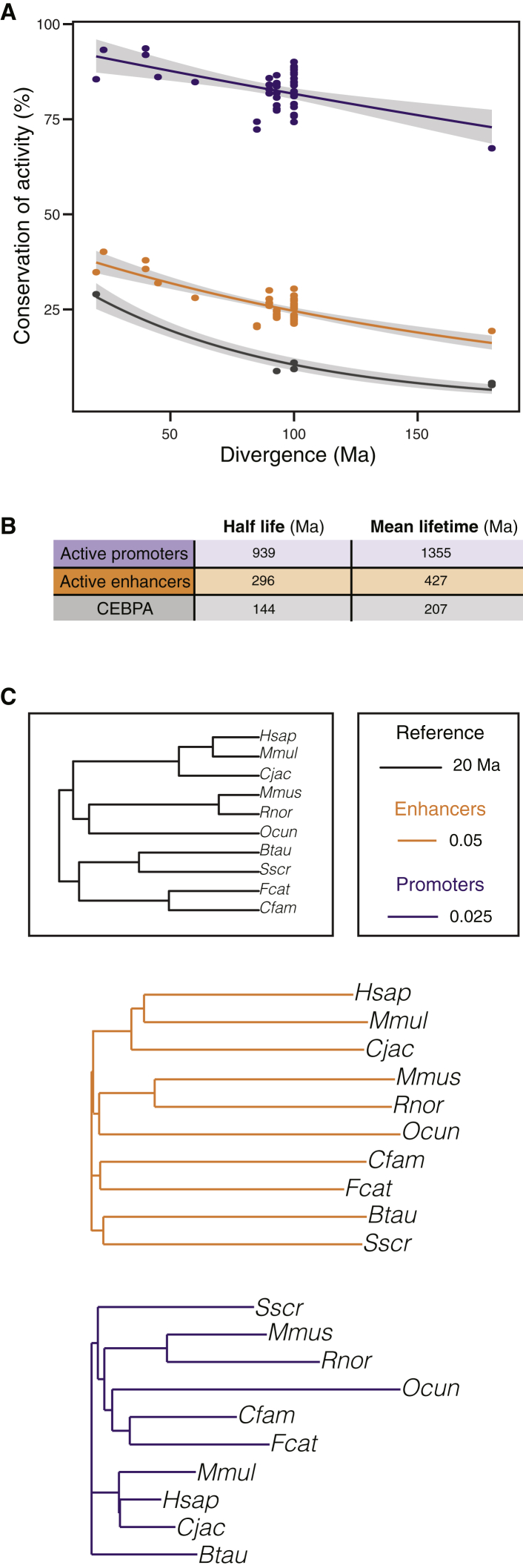
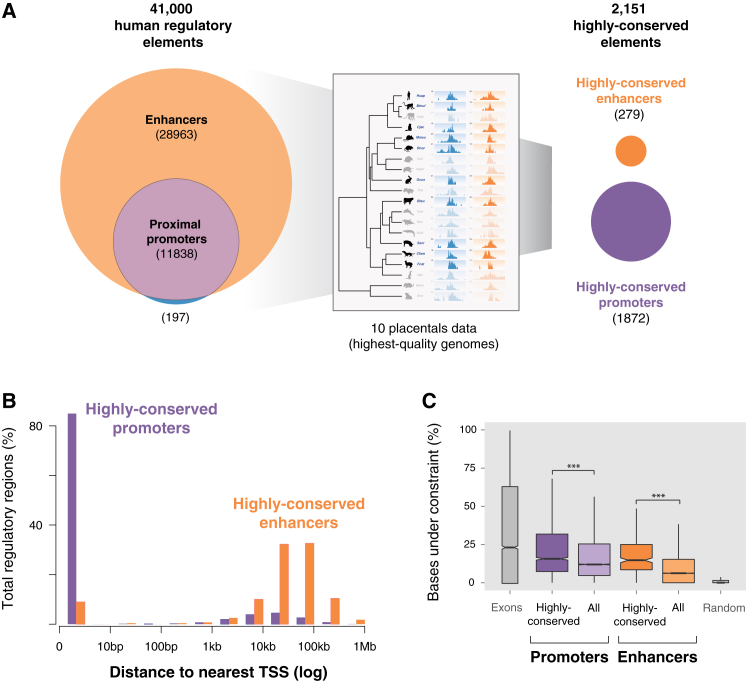
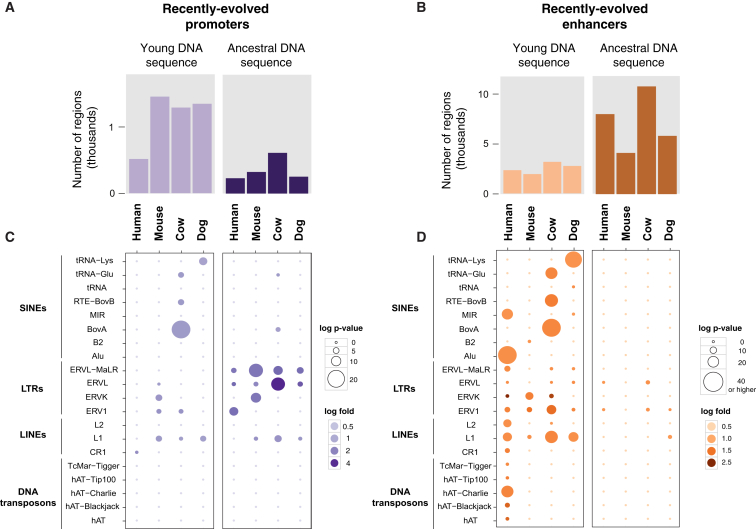
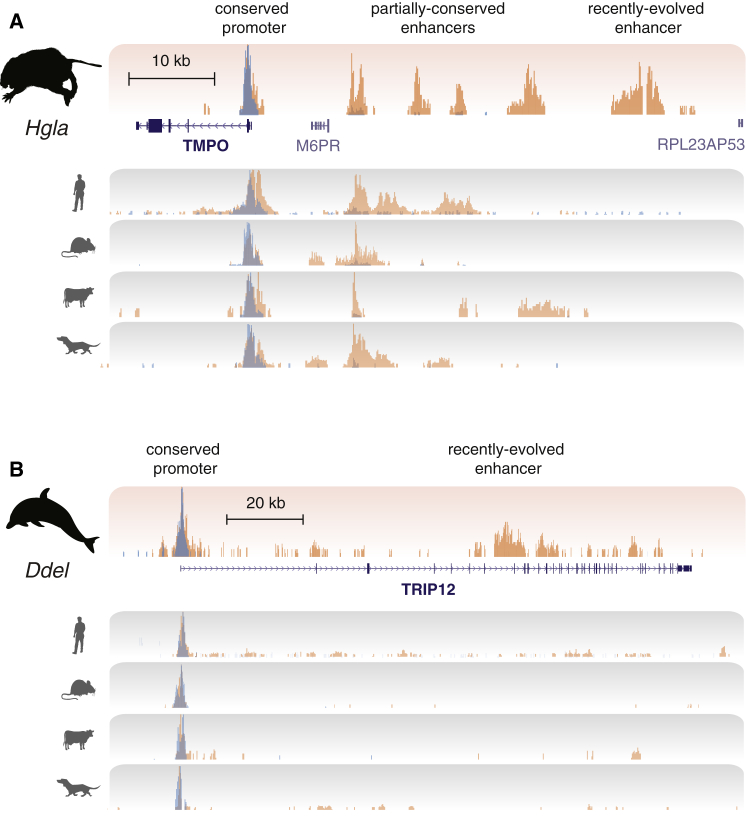
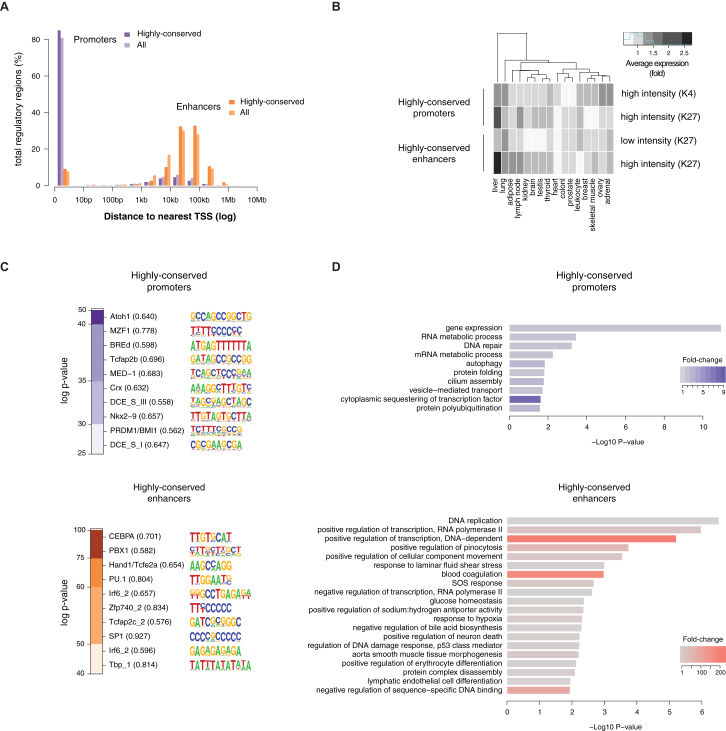
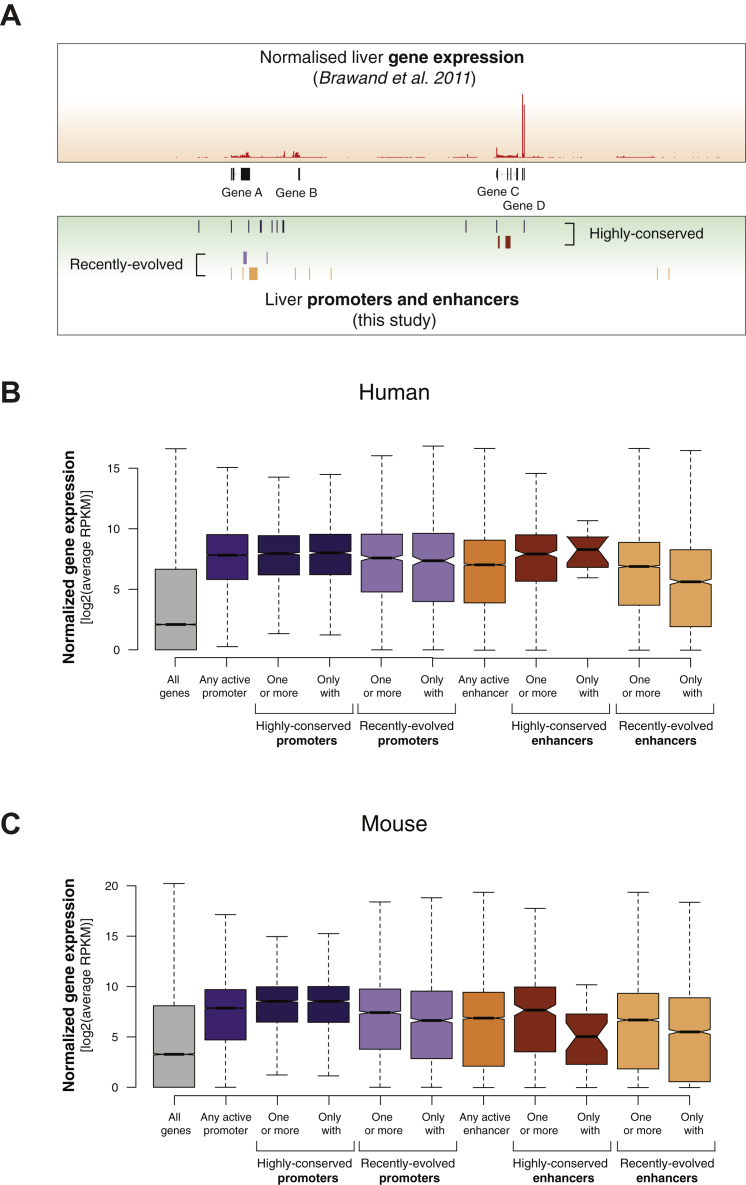
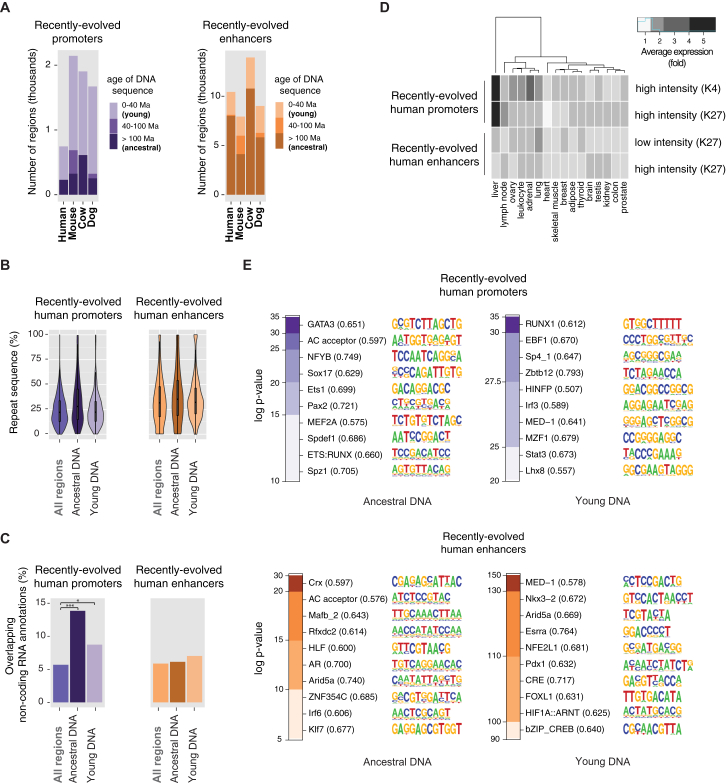
The mammalian radiation has corresponded with rapid changes in noncoding regions of the genome, but we lack a comprehensive understanding of regulatory evolution in mammals. Here, we track the evolution of promoters and enhancers active in liver across 20 mammalian species from six diverse orders by profiling genomic enrichment of H3K27 acetylation and H3K4 trimethylation. We report that rapid evolution of enhancers is a universal feature of mammalian genomes. Most of the recently evolved enhancers arise from ancestral DNA exaptation, rather than lineage-specific expansions of repeat elements. In contrast, almost all liver promoters are partially or fully conserved across these species. Our data further reveal that recently evolved enhancers can be associated with genes under positive selection, demonstrating the power of this approach for annotating regulatory adaptations in genomic sequences. These results provide important insight into the functional genetics underpinning mammalian regulatory evolution.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
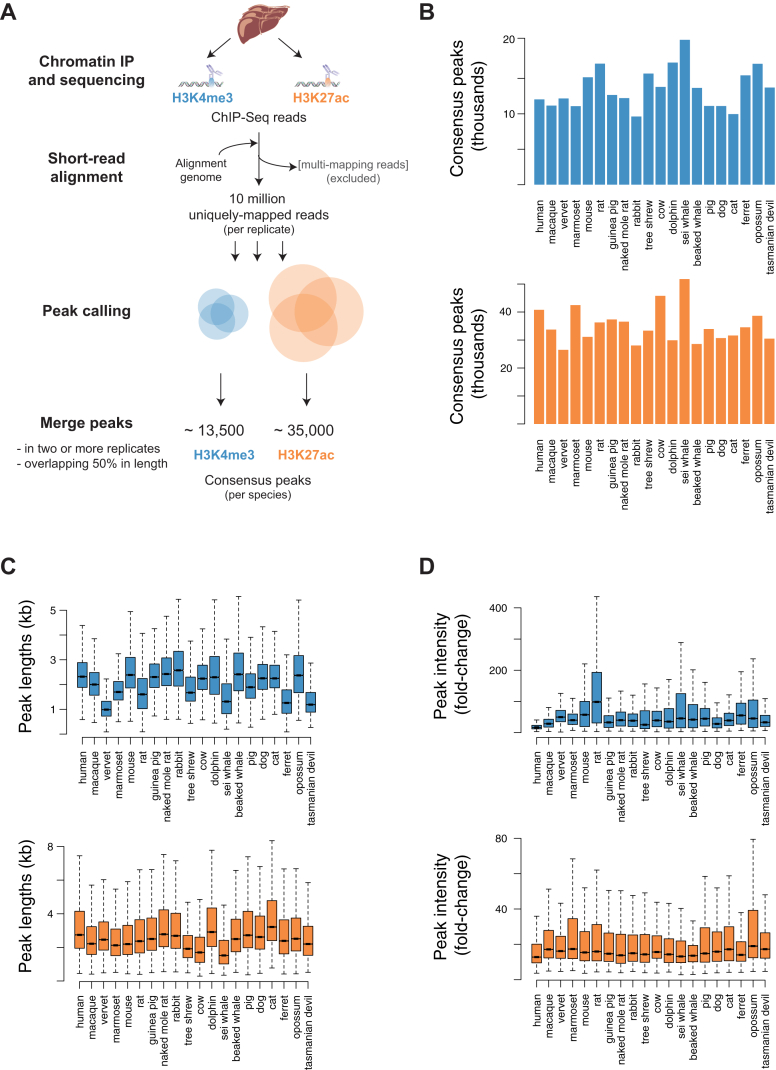
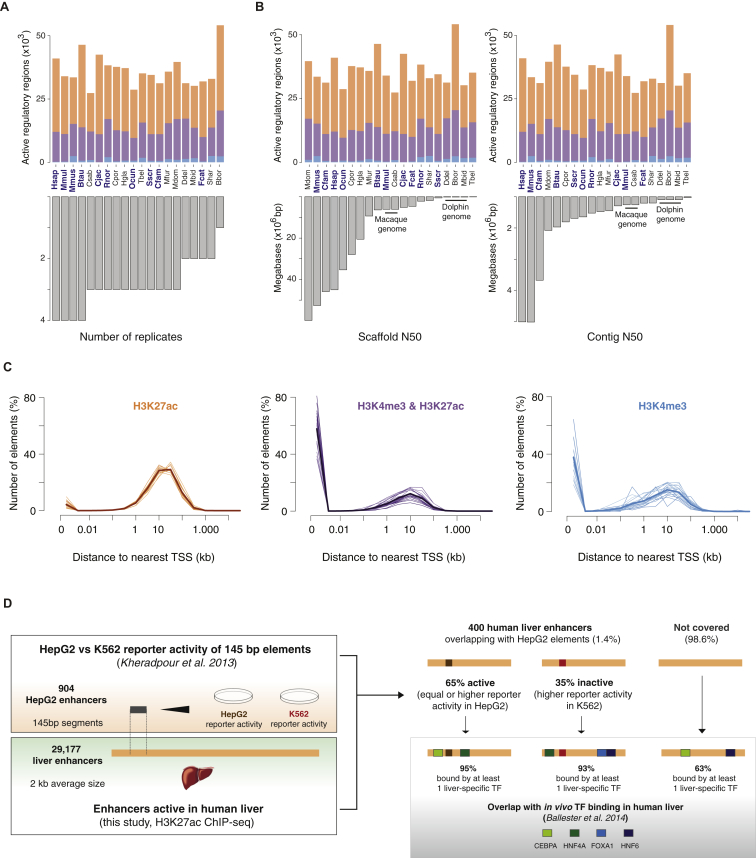
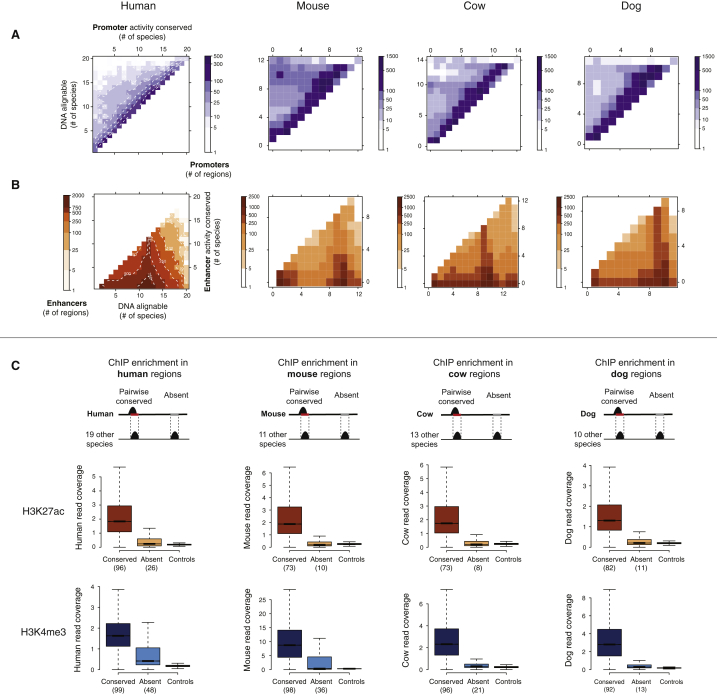
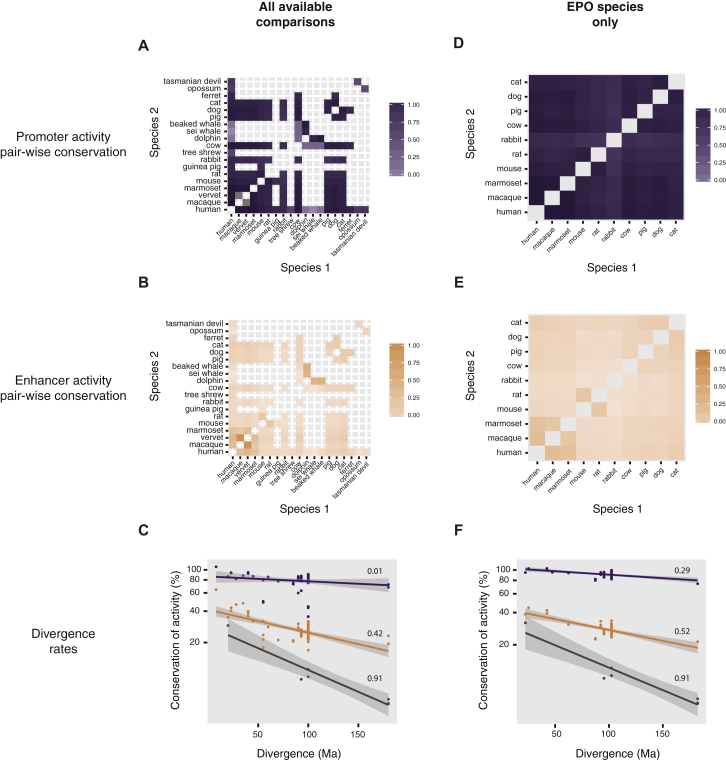
References
Supplemental References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
