

Early detection of structural abnormalities and cytoplasmic accumulation of TDP-43 in tissue-engineered skins derived from ALS patients
- PMID: 25637145
- PMCID: PMC4359444
- DOI: 10.1186/s40478-014-0181-z
Early detection of structural abnormalities and cytoplasmic accumulation of TDP-43 in tissue-engineered skins derived from ALS patients
Abstract
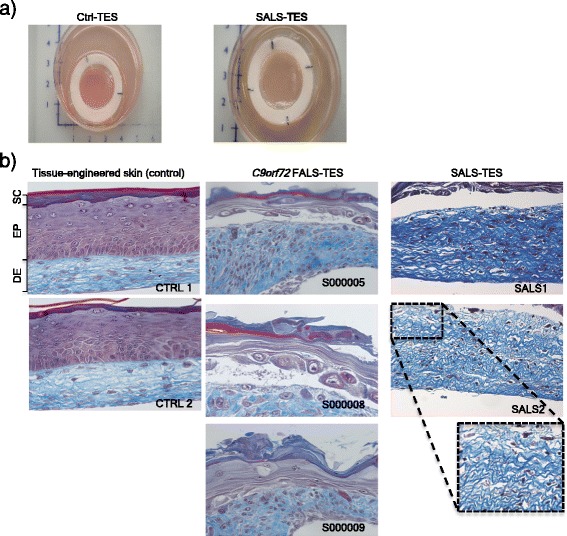
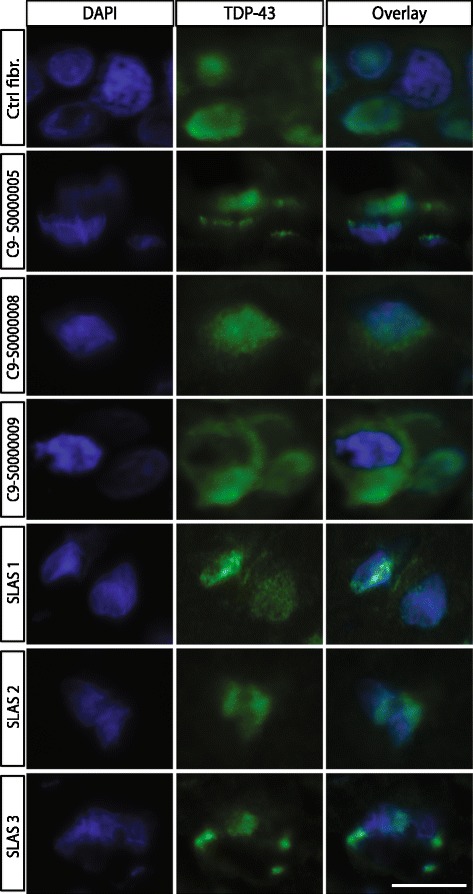
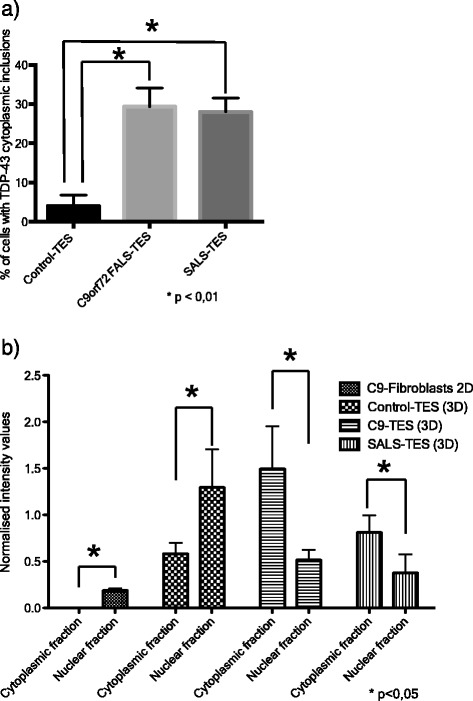
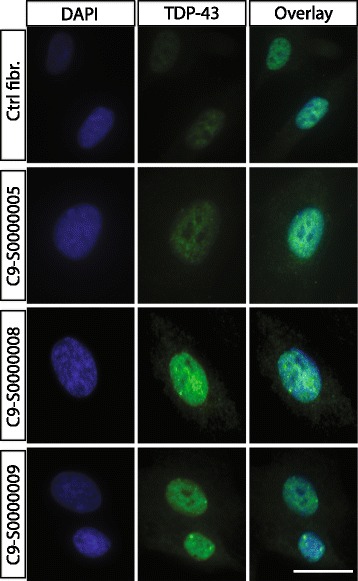
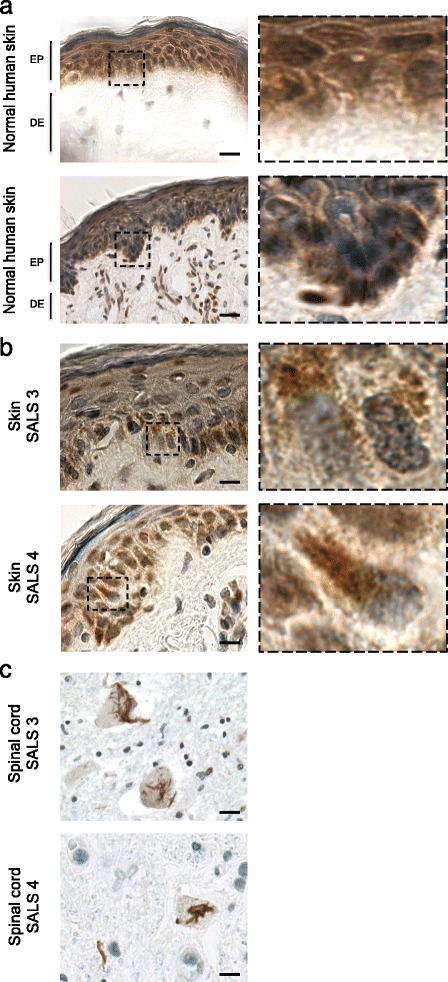
Amyotrophic lateral sclerosis (ALS) is an adult-onset disease characterized by the selective degeneration of motor neurons in the brain and spinal cord progressively leading to paralysis and death. Current diagnosis of ALS is based on clinical assessment of related symptoms. The clinical manifestations observed in ALS appear relatively late in the disease course after degeneration of a significant number of motor neurons. As a result, the identification and development of disease-modifying therapies is difficult. Therefore, novel strategies for early diagnosis of neurodegeneration, to monitor disease progression and to assess response to existing and future treatments are urgently needed. Factually, many neurological disorders, including ALS, are accompanied by skin changes that often precede the onset of neurological symptoms. Aiming to generate an innovative human-based model to facilitate the identification of predictive biomarkers associated with the disease, we developed a unique ALS tissue-engineered skin model (ALS-TES) derived from patient's own cells. The ALS-TES presents a number of striking features including altered epidermal differentiation, abnormal dermo-epidermal junction, delamination, keratinocyte infiltration, collagen disorganization and cytoplasmic TDP-43 inclusions. Remarkably, these abnormal skin defects, uniquely seen in the ALS-derived skins, were detected in pre-symtomatic C9orf72-linked ALS patients carrying the GGGGCC DNA repeat expansion. Consequently, our ALS skin model could represent a renewable source of human tissue, quickly and easily accessible to better understand the physiophatological mechanisms underlying this disease, to facilitate the identification of disease-specific biomarkers, and to develop innovative tools for early diagnosis and disease monitoring.
Figures
References
-
- Brettschneider J, Van Deerlin VM, Robinson JL, Kwong L, Lee EB, Ali YO, Safren N, Monteiro MJ, Toledo JB, Elman L, McCluskey L, Irwin DJ, Grossman M, Molina-Porcel L, Lee VM, Trojanowski JQ. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012;123(6):825–839. doi: 10.1007/s00401-012-0970-z. - DOI - PMC - PubMed
-
- Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, Martindale J, Hartley J, Walsh T, Gelsthorpe C, Baxter L, Forster G, Fox M, Bury J, Mok K, McDermott CJ, Traynor BJ, Kirby J, Wharton SB, Ince PG, Hardy J, Shaw PJ. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135(Pt 3):751–764. doi: 10.1093/brain/awr365. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
