

Identifying anti-growth factors for human cancer cell lines through genome-scale metabolic modeling
- PMID: 25640694
- PMCID: PMC4313100
- DOI: 10.1038/srep08183
Identifying anti-growth factors for human cancer cell lines through genome-scale metabolic modeling
Abstract
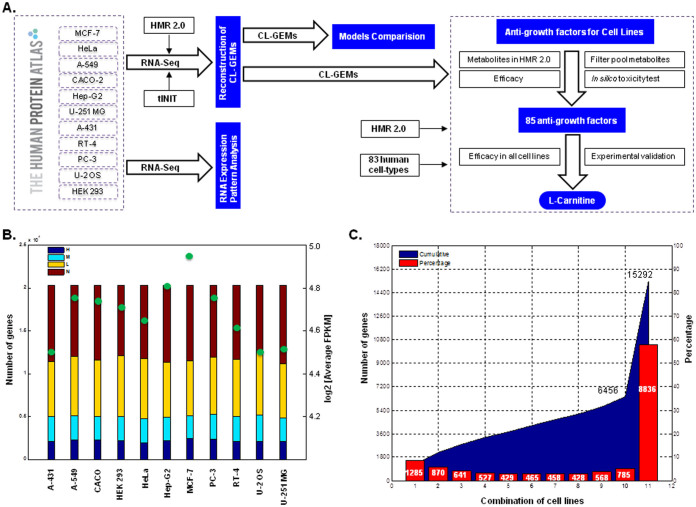
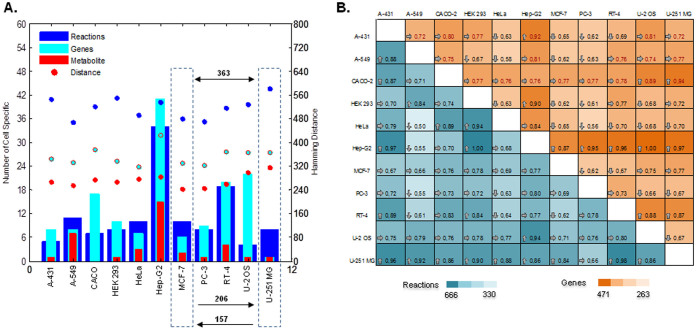
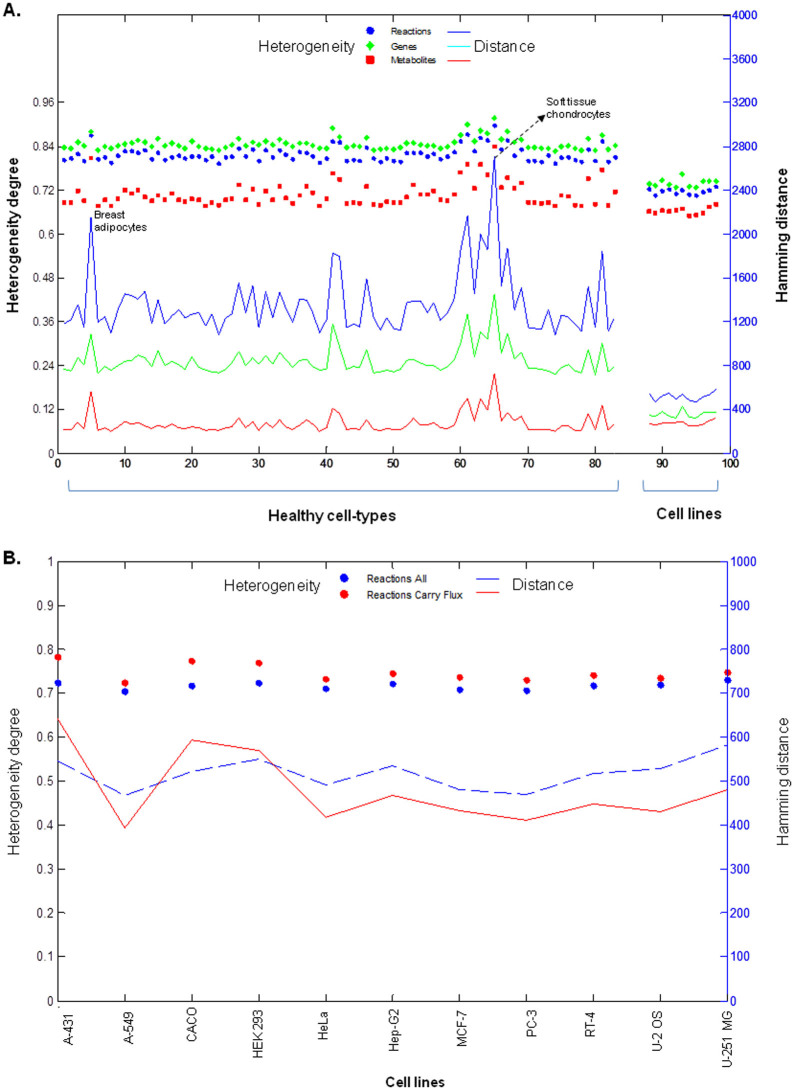
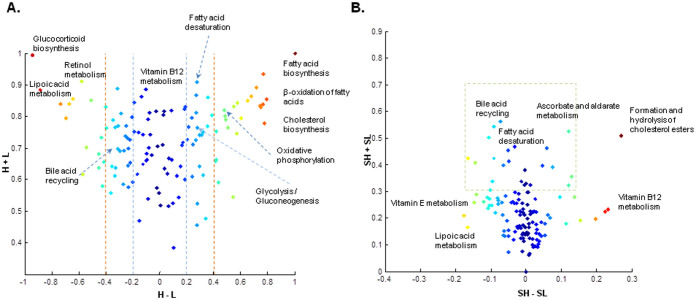
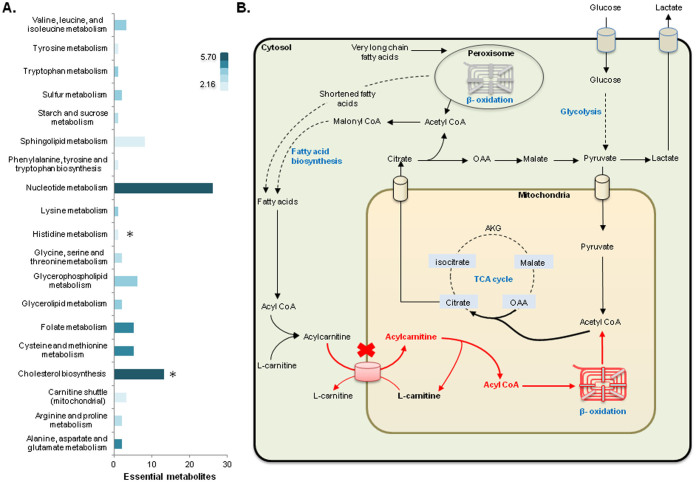
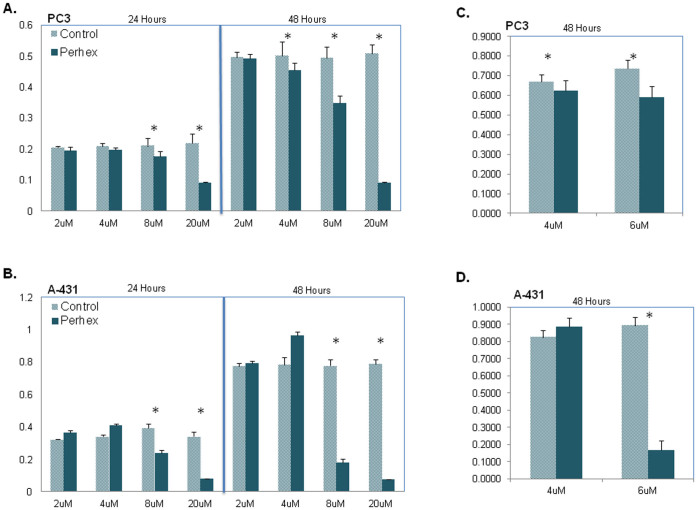
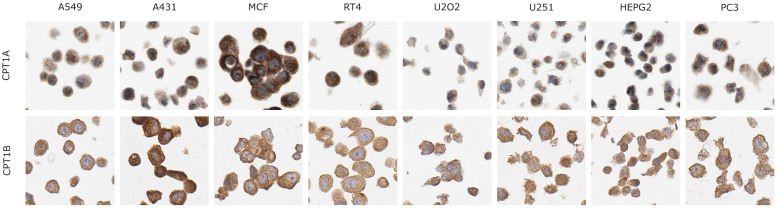
Human cancer cell lines are used as important model systems to study molecular mechanisms associated with tumor growth, hereunder how genomic and biological heterogeneity found in primary tumors affect cellular phenotypes. We reconstructed Genome scale metabolic models (GEMs) for eleven cell lines based on RNA-Seq data and validated the functionality of these models with data from metabolite profiling. We used cell line-specific GEMs to analyze the differences in the metabolism of cancer cell lines, and to explore the heterogeneous expression of the metabolic subsystems. Furthermore, we predicted 85 antimetabolites that can inhibit growth of, or even kill, any of the cell lines, while at the same time not being toxic for 83 different healthy human cell types. 60 of these antimetabolites were found to inhibit growth in all cell lines. Finally, we experimentally validated one of the predicted antimetabolites using two cell lines with different phenotypic origins, and found that it is effective in inhibiting the growth of these cell lines. Using immunohistochemistry, we also showed high or moderate expression levels of proteins targeted by the validated antimetabolite. Identified anti-growth factors for inhibition of cell growth may provide leads for the development of efficient cancer treatment strategies.
Figures
Similar articles
-
Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling.Mol Syst Biol. 2014 Mar 19;10(3):721. doi: 10.1002/msb.145122. Mol Syst Biol. 2014. PMID: 24646661 Free PMC article.
-
Inhibition of long-chain fatty acid metabolism does not affect platelet aggregation responses.Eur J Pharmacol. 1998 Sep 4;356(2-3):207-13. doi: 10.1016/s0014-2999(98)00527-5. Eur J Pharmacol. 1998. PMID: 9774251
-
Identification of Cancer-associated metabolic vulnerabilities by modeling multi-objective optimality in metabolism.Cell Commun Signal. 2019 Oct 10;17(1):124. doi: 10.1186/s12964-019-0439-y. Cell Commun Signal. 2019. PMID: 31601242 Free PMC article.
-
Genome-scale modeling of human metabolism - a systems biology approach.Biotechnol J. 2013 Sep;8(9):985-96. doi: 10.1002/biot.201200275. Epub 2013 Apr 24. Biotechnol J. 2013. PMID: 23613448 Review.
-
New paradigms for metabolic modeling of human cells.Curr Opin Biotechnol. 2015 Aug;34:91-7. doi: 10.1016/j.copbio.2014.12.013. Epub 2015 Jan 2. Curr Opin Biotechnol. 2015. PMID: 25559199 Review.
Cited by
-
Reconstruction and validation of a constraint-based metabolic network model for bone marrow-derived mesenchymal stem cells.Cell Prolif. 2015 Aug;48(4):475-85. doi: 10.1111/cpr.12197. Epub 2015 Jul 1. Cell Prolif. 2015. PMID: 26132591 Free PMC article.
-
Current State of "Omics" Biomarkers in Pancreatic Cancer.J Pers Med. 2021 Feb 14;11(2):127. doi: 10.3390/jpm11020127. J Pers Med. 2021. PMID: 33672926 Free PMC article. Review.
-
Reconstruction of Tissue-Specific Metabolic Networks Using CORDA.PLoS Comput Biol. 2016 Mar 4;12(3):e1004808. doi: 10.1371/journal.pcbi.1004808. eCollection 2016 Mar. PLoS Comput Biol. 2016. PMID: 26942765 Free PMC article.
-
Host-mycobiome metabolic interactions in health and disease.Gut Microbes. 2022 Jan-Dec;14(1):2121576. doi: 10.1080/19490976.2022.2121576. Gut Microbes. 2022. PMID: 36151873 Free PMC article.
-
Personalized Cardiovascular Disease Prediction and Treatment-A Review of Existing Strategies and Novel Systems Medicine Tools.Front Physiol. 2016 Jan 26;7:2. doi: 10.3389/fphys.2016.00002. eCollection 2016. Front Physiol. 2016. PMID: 26858650 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
