

Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow
- PMID: 25641345
- PMCID: PMC4404517
- DOI: 10.1111/micc.12190
Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow
Abstract
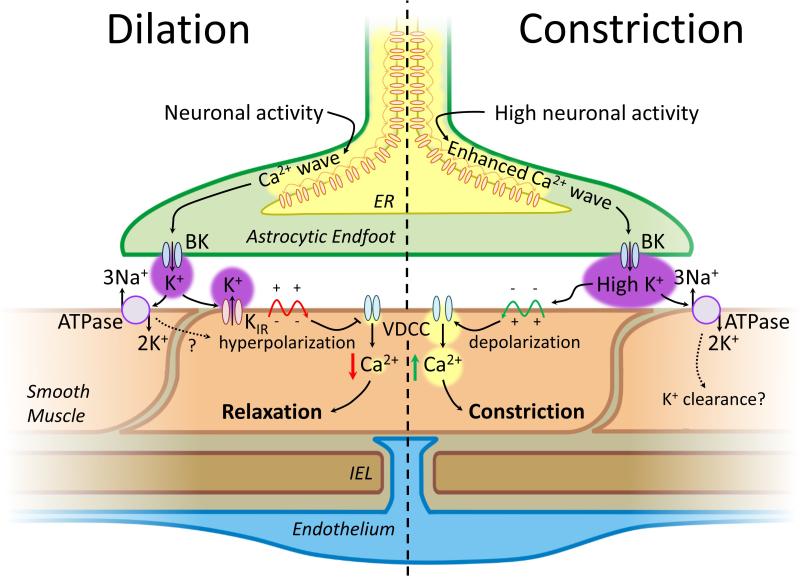
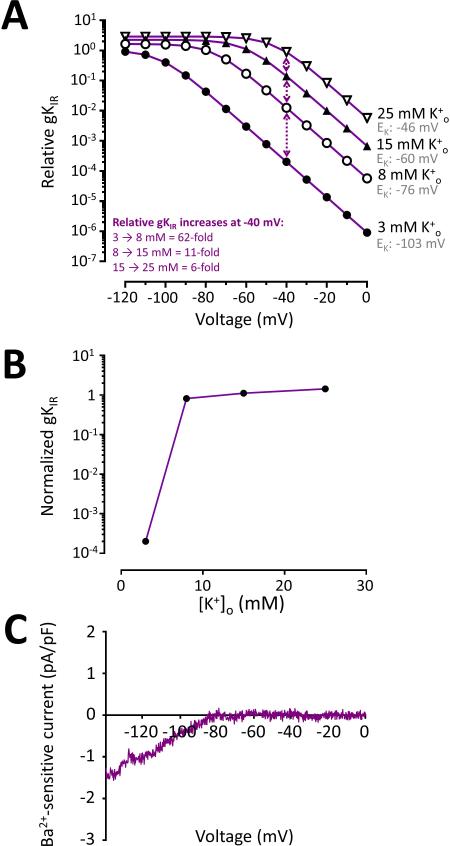
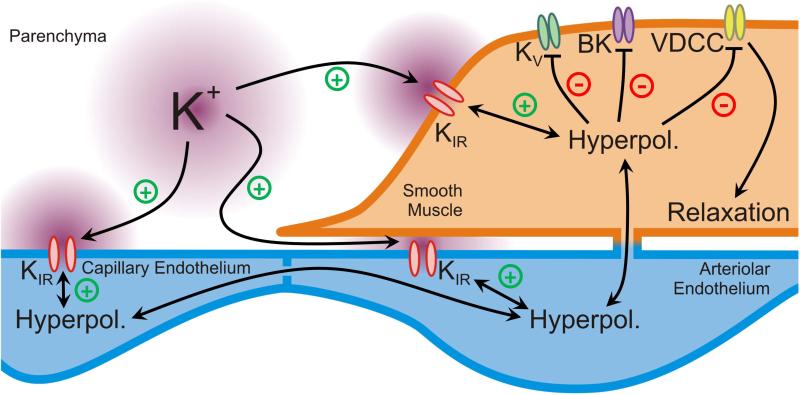
For decades it has been known that external K(+) ions are rapid and potent vasodilators that increase CBF. Recent studies have implicated the local release of K(+) from astrocytic endfeet-which encase the entirety of the parenchymal vasculature-in the dynamic regulation of local CBF during NVC. It has been proposed that the activation of KIR channels in the vascular wall by external K(+) is a central component of these hyperemic responses; however, a number of significant gaps in our knowledge remain. Here, we explore the concept that vascular KIR channels are the major extracellular K(+) sensors in the control of CBF. We propose that K(+) is an ideal mediator of NVC, and discuss KIR channels as effectors that produce rapid hyperpolarization and robust vasodilation of cerebral arterioles. We provide evidence that KIR channels, of the KIR 2 subtype in particular, are present in both the endothelial and SM cells of parenchymal arterioles and propose that this dual positioning of KIR 2 channels increases the robustness of the vasodilation to external K(+), enables the endothelium to be actively engaged in NVC, and permits electrical signaling through the endothelial syncytium to promote upstream vasodilation to modulate CBF.
Keywords: astrocytic endfoot; capillary; cerebral blood flow; endothelium; functional hyperemia; inward rectifier potassium channel; neurovascular coupling; parenchymal arteriole; smooth muscle.
© 2015 John Wiley & Sons Ltd.
Figures

References
-
- Back T, Kohno K, Hossmann KA. Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression: effect on blood flow, tissue oxygenation, and electroencephalogram. J Cereb Blood Flow Metab. 1994;14:12–19. - PubMed
-
- Bhandari S, Hunter M. Biophysical effects of pore mutations of ROMK1. Clin Sci. 2001;101:121–30. - PubMed
-
- Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
