

Efficient Bayesian mixed-model analysis increases association power in large cohorts
- PMID: 25642633
- PMCID: PMC4342297
- DOI: 10.1038/ng.3190
Efficient Bayesian mixed-model analysis increases association power in large cohorts
Abstract
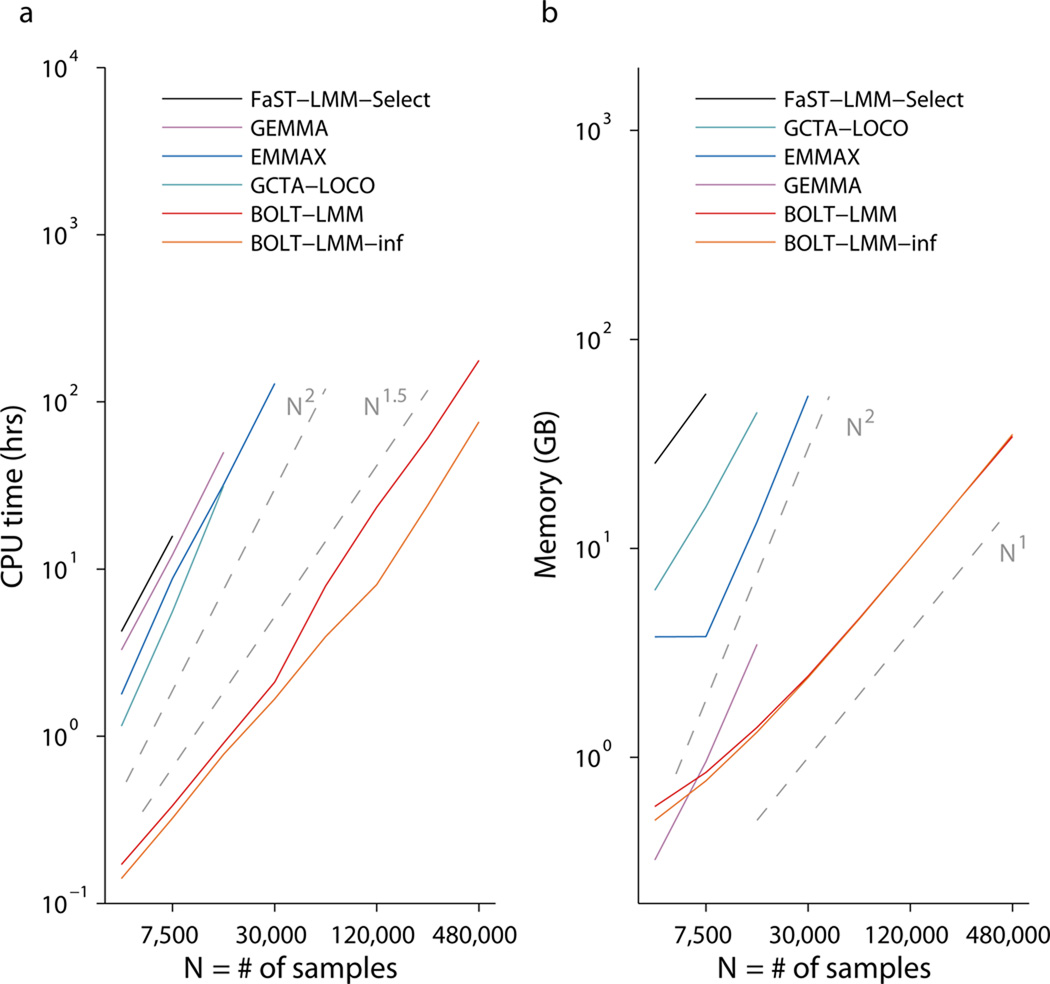
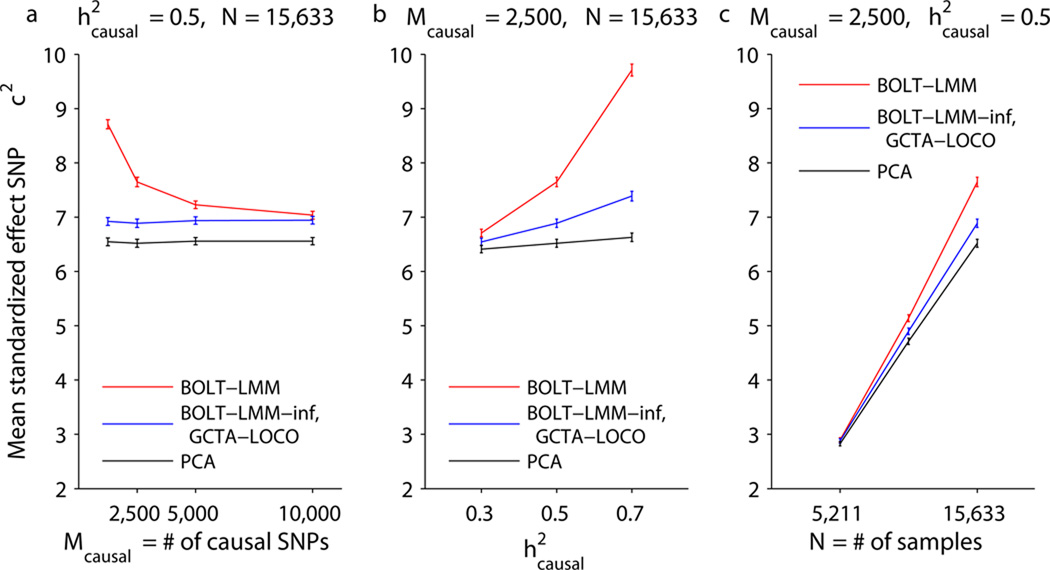
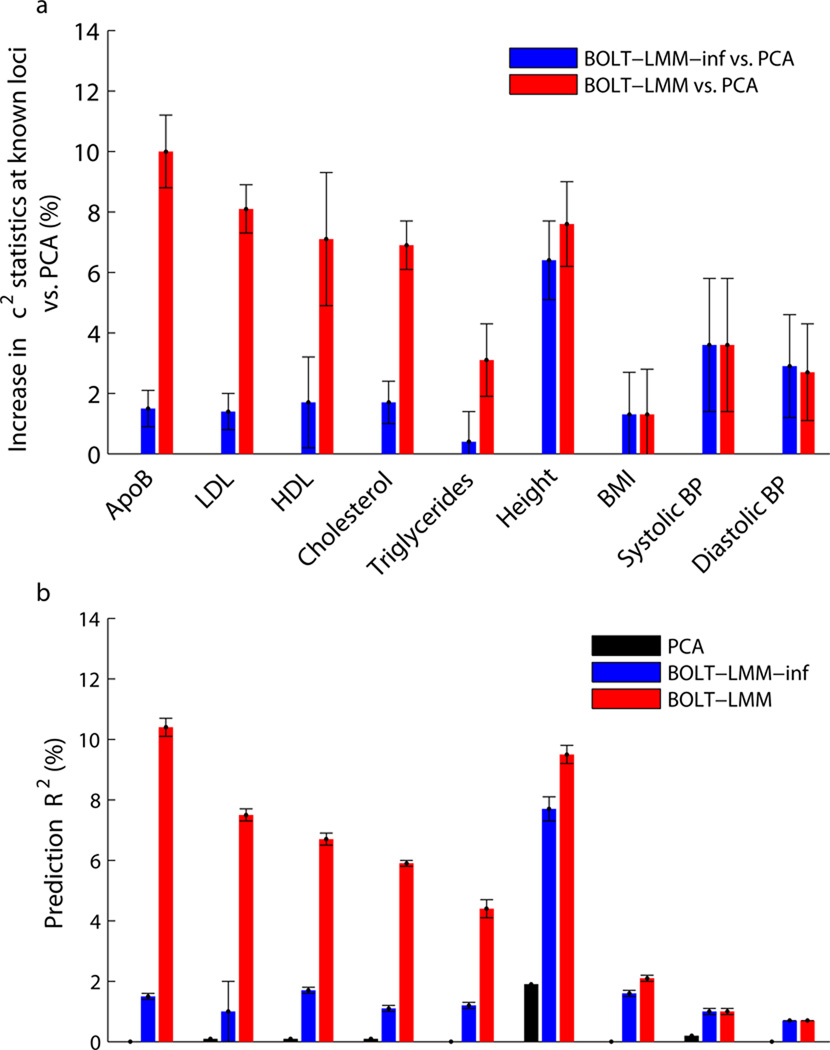
Linear mixed models are a powerful statistical tool for identifying genetic associations and avoiding confounding. However, existing methods are computationally intractable in large cohorts and may not optimize power. All existing methods require time cost O(MN(2)) (where N is the number of samples and M is the number of SNPs) and implicitly assume an infinitesimal genetic architecture in which effect sizes are normally distributed, which can limit power. Here we present a far more efficient mixed-model association method, BOLT-LMM, which requires only a small number of O(MN) time iterations and increases power by modeling more realistic, non-infinitesimal genetic architectures via a Bayesian mixture prior on marker effect sizes. We applied BOLT-LMM to 9 quantitative traits in 23,294 samples from the Women's Genome Health Study (WGHS) and observed significant increases in power, consistent with simulations. Theory and simulations show that the boost in power increases with cohort size, making BOLT-LMM appealing for genome-wide association studies in large cohorts.
Figures
References
References (Online Methods)
-
- Boyd SP, Vandenberghe L. Convex Optimization. Cambridge University Press; 2004.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
