

Structural basis for the inhibition of M1 family aminopeptidases by the natural product actinonin: Crystal structure in complex with E. coli aminopeptidase N
- PMID: 25644575
- PMCID: PMC4420530
- DOI: 10.1002/pro.2653
Structural basis for the inhibition of M1 family aminopeptidases by the natural product actinonin: Crystal structure in complex with E. coli aminopeptidase N
Abstract
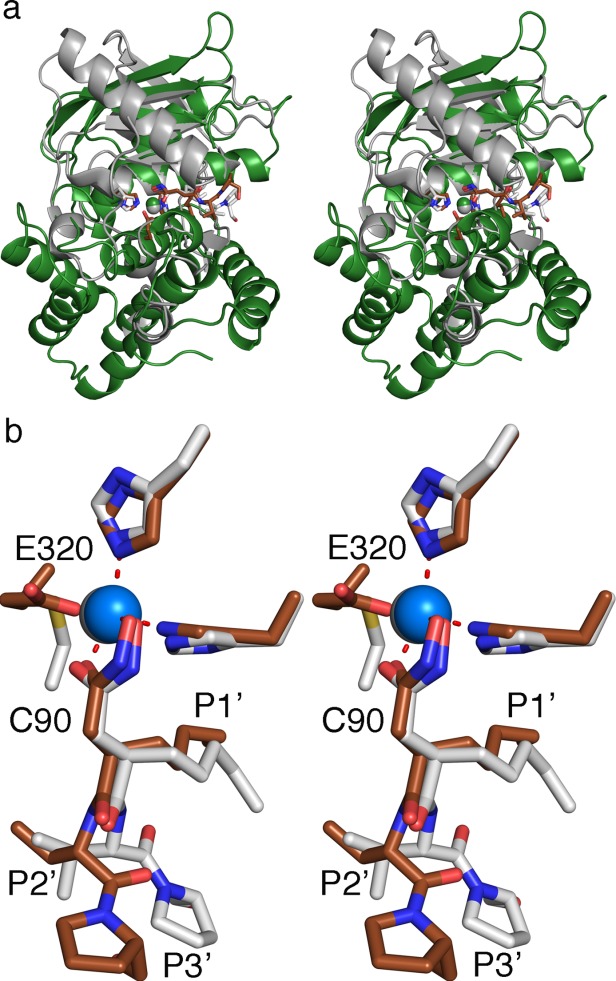
Actinonin is a pseudotripeptide that displays a high affinity towards metalloproteases including peptide deformylases (PDFs) and M1 family aminopeptidases. PDF and M1 family aminopeptidases belong to thermolysin-metzincin superfamily. One of the major differences in terms of substrate binding pockets between these families is presence (in M1 aminopeptidases) or absence (in PDFs) of an S1 substrate pocket. The binding mode of actinonin to PDFs has been established previously; however, it is not clear how the actinonin, without a P1 residue, would bind to the M1 aminopeptidases. Here we describe the crystal structure of Escherichia coli aminopeptidase N (ePepN), a model protein of the M1 family aminopeptidases in complex with actinonin. For comparison we have also determined the structure of ePepN in complex with a well-known tetrapeptide inhibitor, amastatin. From the comparison of the actinonin and amastatin ePepN complexes, it is clear that the P1 residue is not critical as long as strong metal chelating head groups, like hydroxamic acid or α-hydroxy ketone, are present. Results from this study will be useful for the design of selective and efficient hydroxamate inhibitors against M1 family aminopeptidases.
Keywords: M1 aminopeptidase; actinonin; amastatin; matrix metalloproteinase; peptide deformylase.
© 2015 The Protein Society.
Figures




References
-
- Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, Margolis PS, Wu C, Ni ZJ, Trias J, White RJ, Yuan Z. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry. 2000;39:1256–1262. - PubMed
-
- Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline in 2013. J Antibiot. 2013;66:571–591. - PubMed
-
- Lee MD, Antczak C, Li Y, Sirotnak FM, Bornmann WG, Scheinberg DA. A new human peptide deformylase inhibitable by actinonin. Biochem Biophys Res Commun. 2003;312:309–315. - PubMed
-
- Xu Y, Lai LT, Gabrilove JL, Scheinberg DA. Antitumor activity of actinonin in vitro and in vivo. Clin Cancer Res. 1998;4:171–176. - PubMed
-
- Bernstein FC, Koetzle TF, Williams GJ, Meyer EF., Jr Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
