

Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-κB pathways
- PMID: 25644821
- PMCID: PMC4314638
- DOI: 10.1038/srep08209
Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-κB pathways
Abstract
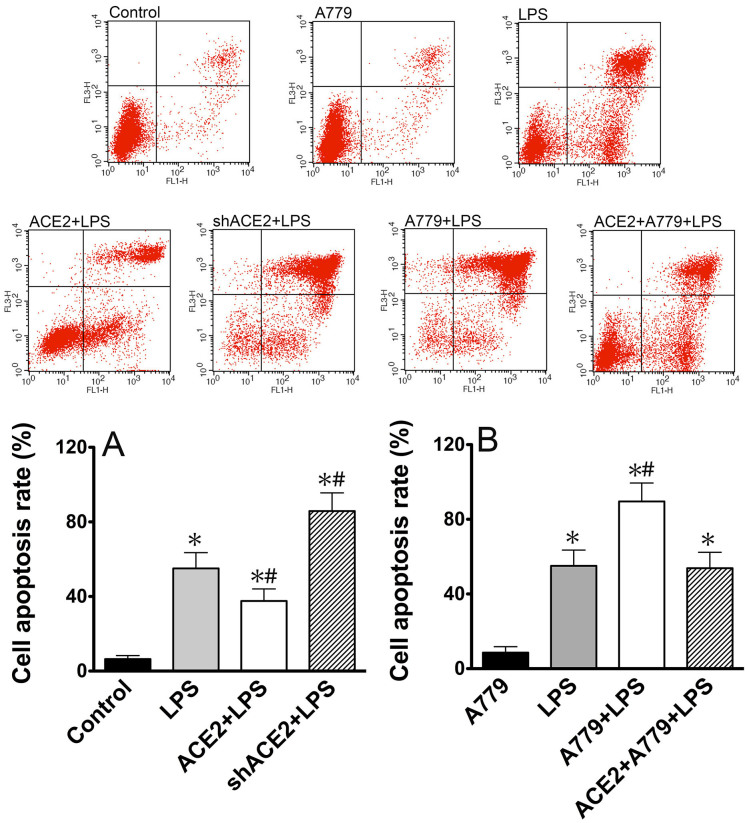
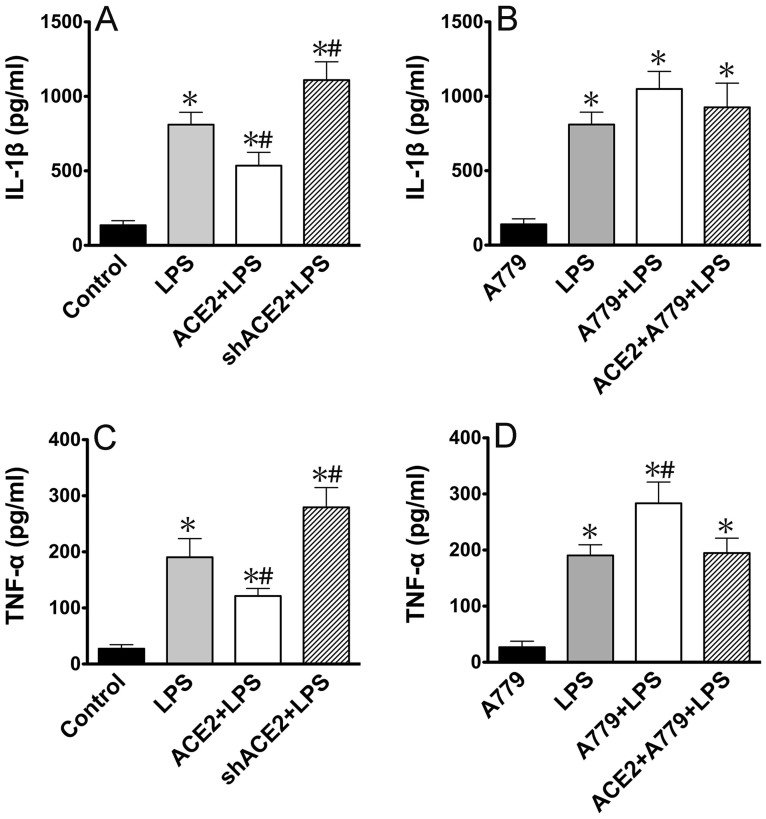
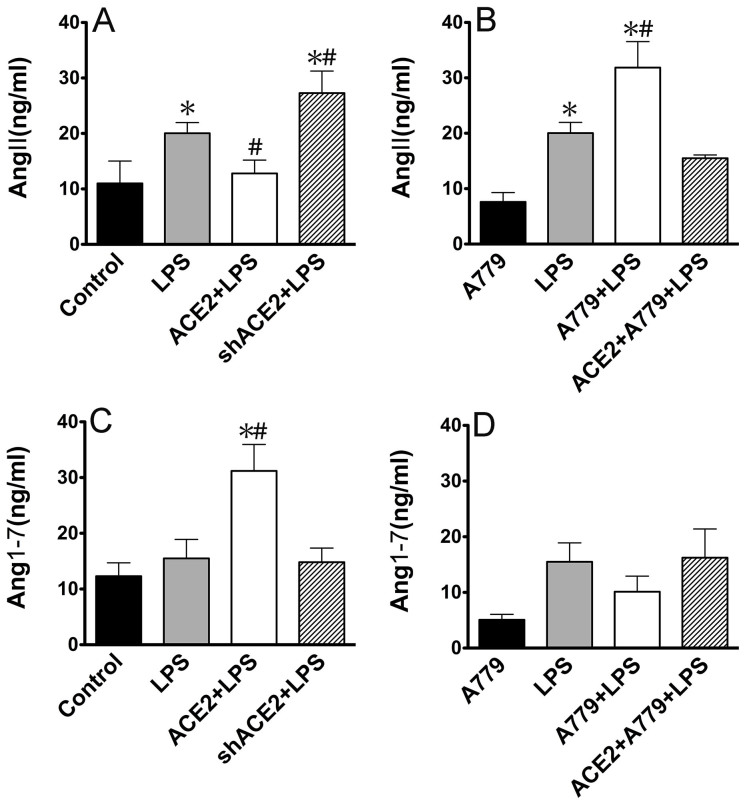
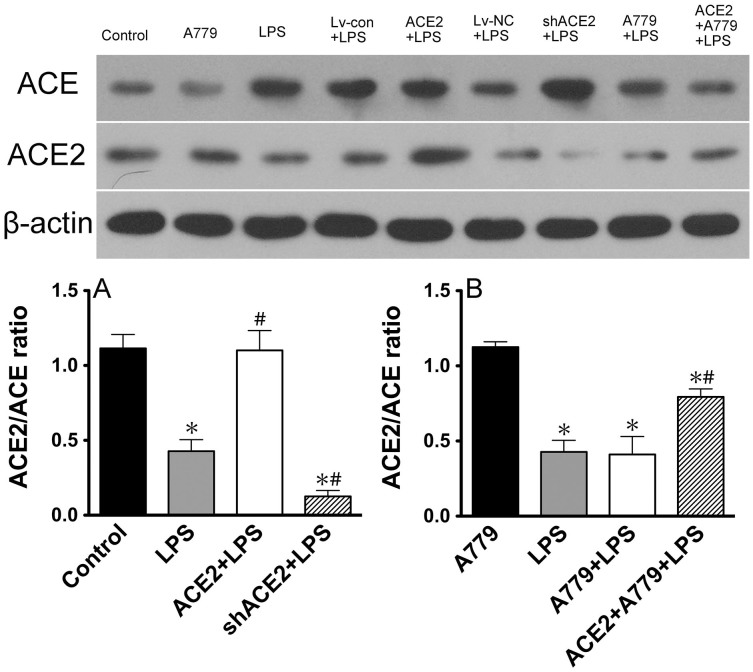
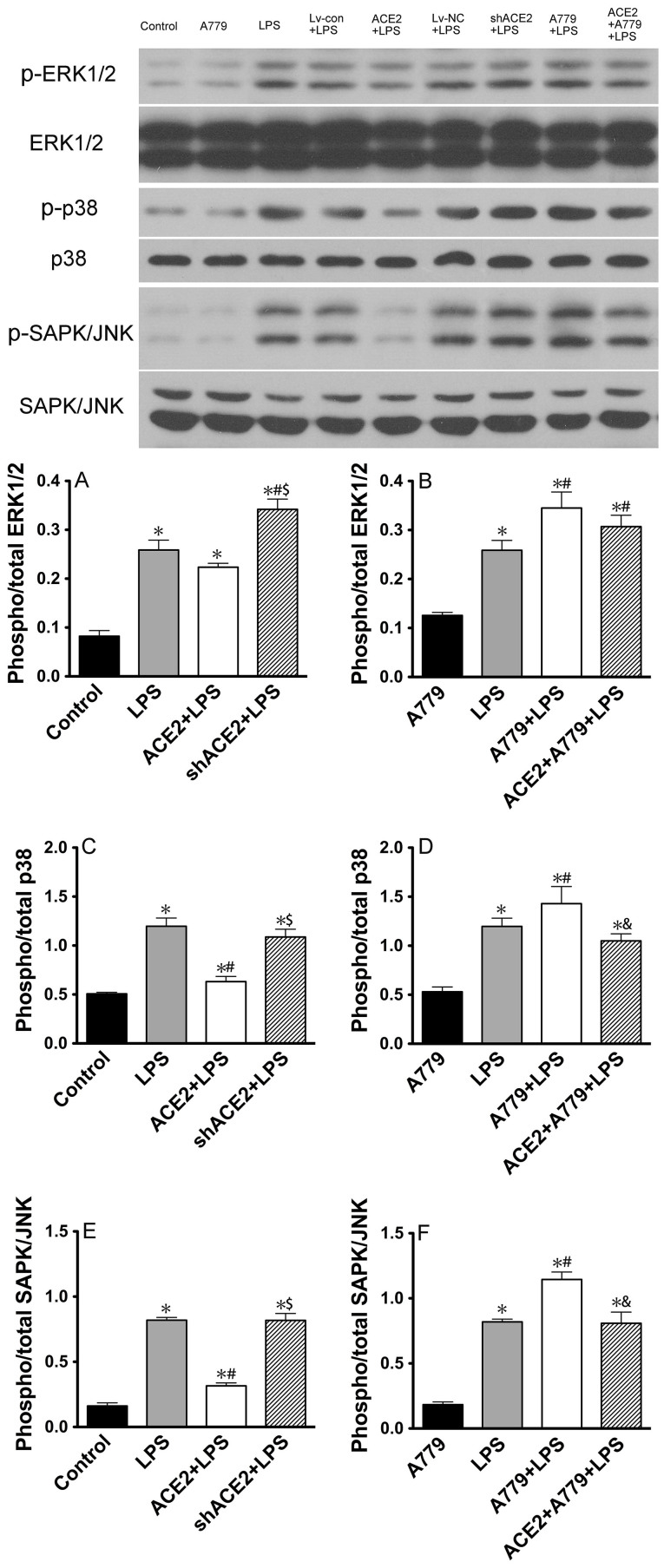
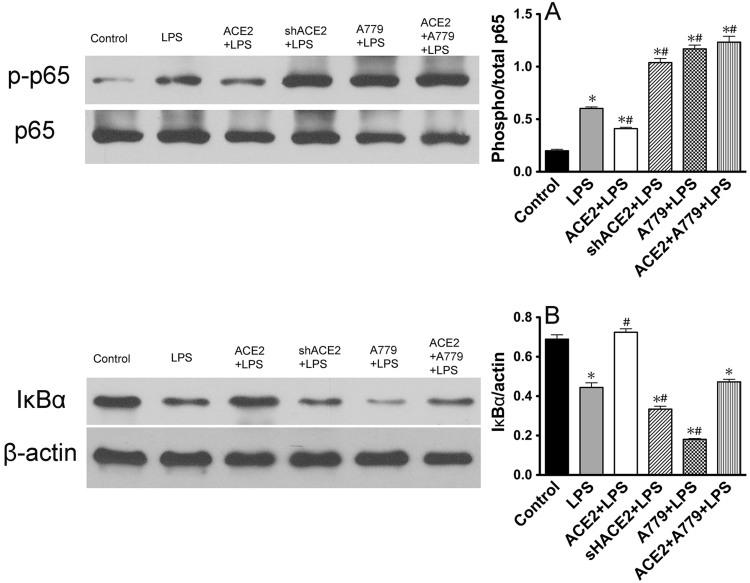
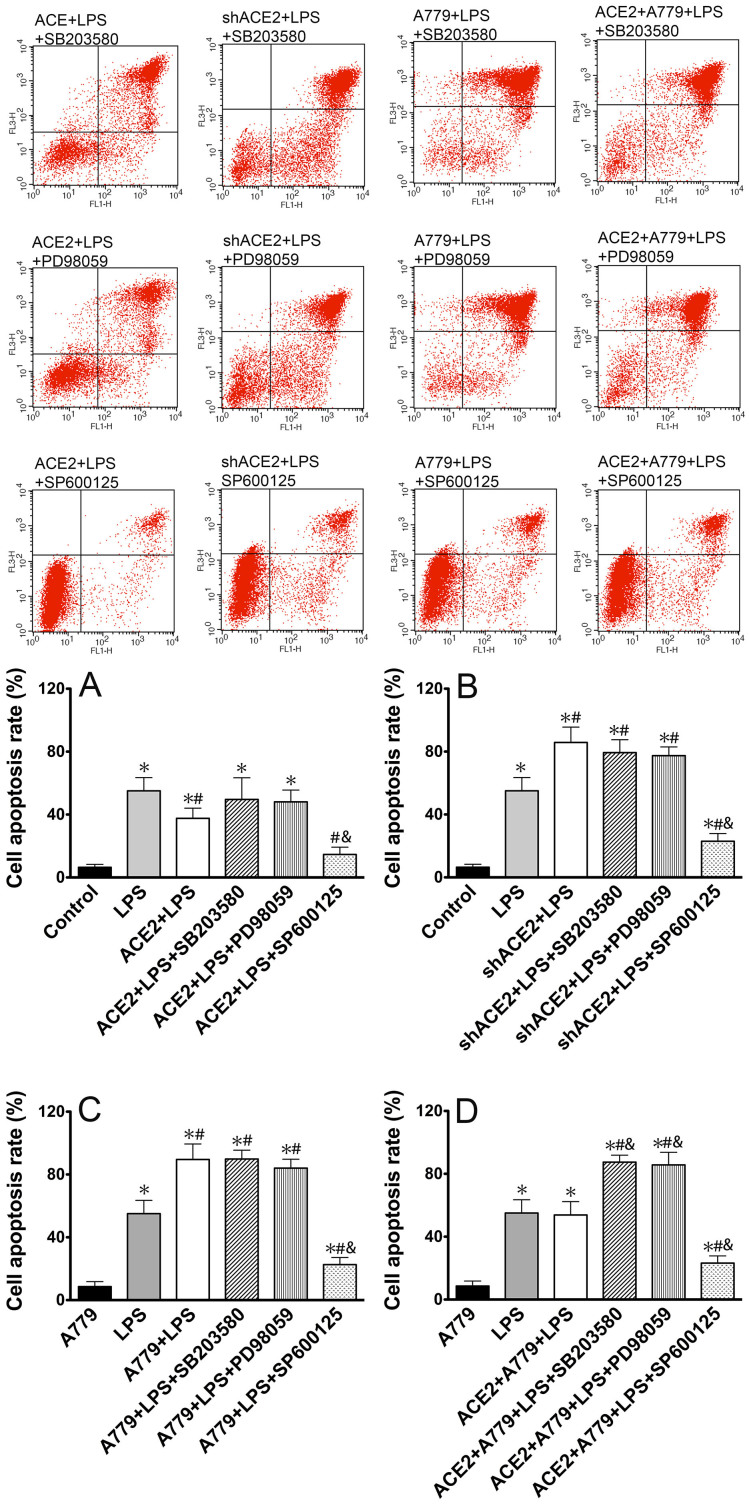
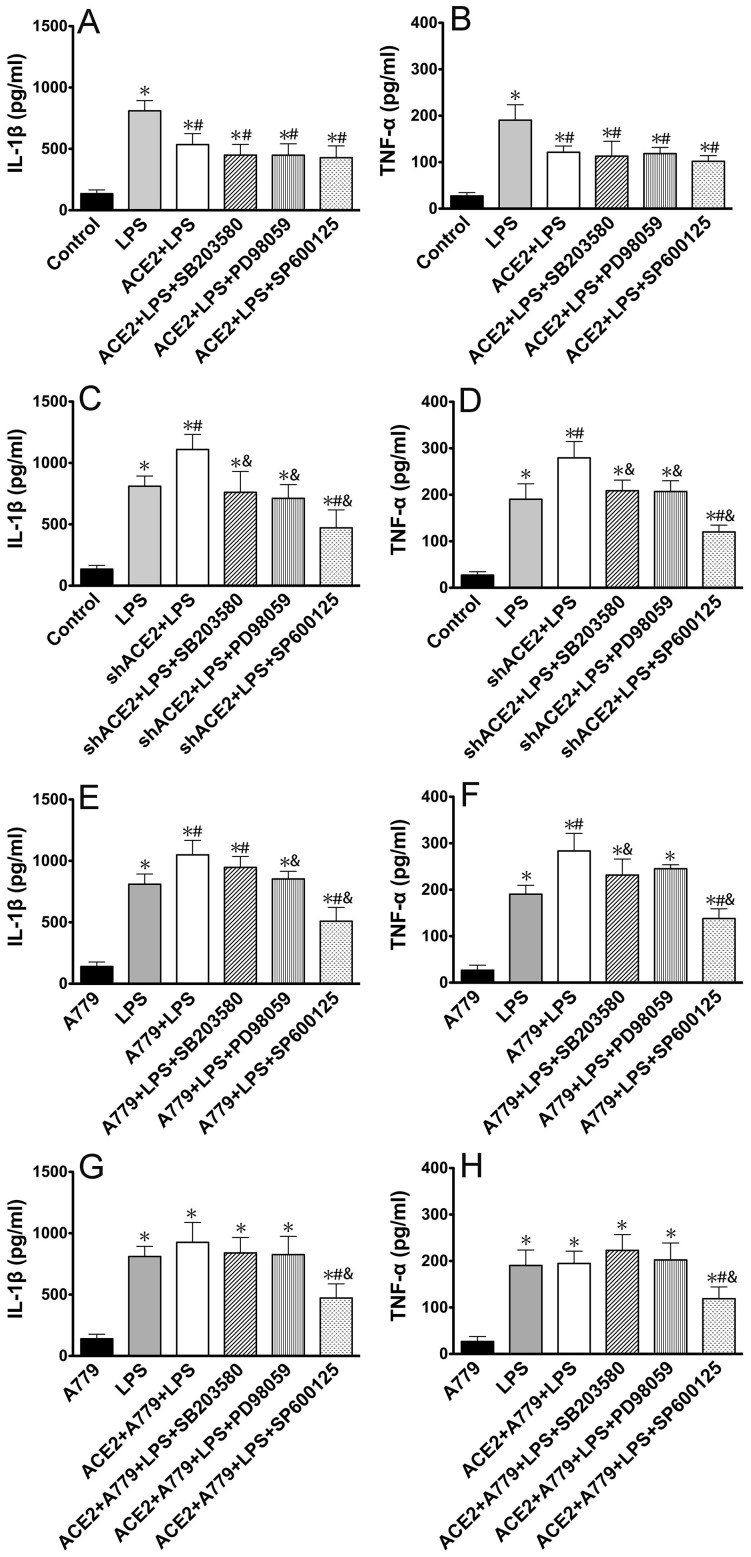
ACE2 and Ang-(1-7) have important roles in preventing acute lung injury. However, it is not clear whether upregulation of the ACE2/Ang-(1-7)/Mas axis prevents LPS-induced injury in pulmonary microvascular endothelial cells (PMVECs) by inhibiting the MAPKs/NF-κB pathways. Primary cultured rat PMVECs were transduced with lentiviral-borne Ace2 or shRNA-Ace2, and then treated or not with Mas receptor blocker (A779) before exposure to LPS. LPS stimulation resulted in the higher levels of AngII, Ang-(1-7), cytokine secretion, and apoptosis rates, and the lower ACE2/ACE ratio. Ace2 reversed the ACE2/ACE imbalance and increased Ang-(1-7) levels, thus reducing LPS-induced apoptosis and inflammation, while inhibition of Ace2 reversed all these effects. A779 abolished these protective effects of Ace2. LPS treatment was associated with activation of the ERK, p38, JNK, and NF-κB pathways, which were aggravated by A779. Pretreatment with A779 prevented the Ace2-induced blockade of p38, JNK, and NF-κB phosphorylation. However, only JNK inhibitor markedly reduced apoptosis and cytokine secretion in PMVECs with Ace2 deletion and A779 pretreatment. These results suggest that the ACE2/Ang-(1-7)/Mas axis has a crucial role in preventing LPS-induced apoptosis and inflammation of PMVECs, by inhibiting the JNK/NF-κB pathways.
Figures
References
-
- Rubenfeld G. D. et al. Incidence and Outcomes of Acute Lung Injury. N Engl J Med 353, 1685–93 (2005). - PubMed
-
- Phua J. et al. Has mortality from acute respiratory distress syndrome decreased over time? : A systematic review. Am J Respir Crit Care Med 179, 220–7 (2009). - PubMed
-
- Cheng C. et al. Lipopolysaccharide induces expression of SSeCKS in rat lung microvascular endothelial cell. Mol Cell Biochem 305, 1–8 (2007). - PubMed
-
- Weir M. R. & Dzau V. J. The renin–angiotensin–aldosterone system: a specific target for hypertension management. Am J Hypertens 12, 205S–213S (1999). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
