

Molecular phylogeny of sequenced Saccharomycetes reveals polyphyly of the alternative yeast codon usage
- PMID: 25646540
- PMCID: PMC4986446
- DOI: 10.1093/gbe/evu152
Molecular phylogeny of sequenced Saccharomycetes reveals polyphyly of the alternative yeast codon usage
Abstract
The universal genetic code defines the translation of nucleotide triplets, called codons, into amino acids. In many Saccharomycetes a unique alteration of this code affects the translation of the CUG codon, which is normally translated as leucine. Most of the species encoding CUG alternatively as serine belong to the Candida genus and were grouped into a so-called CTG clade. However, the "Candida genus" is not a monophyletic group and several Candida species are known to use the standard CUG translation. The codon identity could have been changed in a single branch, the ancestor of the Candida, or to several branches independently leading to a polyphyletic alternative yeast codon usage (AYCU). In order to resolve the monophyly or polyphyly of the AYCU, we performed a phylogenomics analysis of 26 motor and cytoskeletal proteins from 60 sequenced yeast species. By investigating the CUG codon positions with respect to sequence conservation at the respective alignment positions, we were able to unambiguously assign the standard code or AYCU. Quantitative analysis of the highly conserved leucine and serine alignment positions showed that 61.1% and 17% of the CUG codons coding for leucine and serine, respectively, are at highly conserved positions, whereas only 0.6% and 2.3% of the CUG codons, respectively, are at positions conserved in the respective other amino acid. Plotting the codon usage onto the phylogenetic tree revealed the polyphyly of the AYCU with Pachysolen tannophilus and the CTG clade branching independently within a time span of 30–100 Ma.
Figures
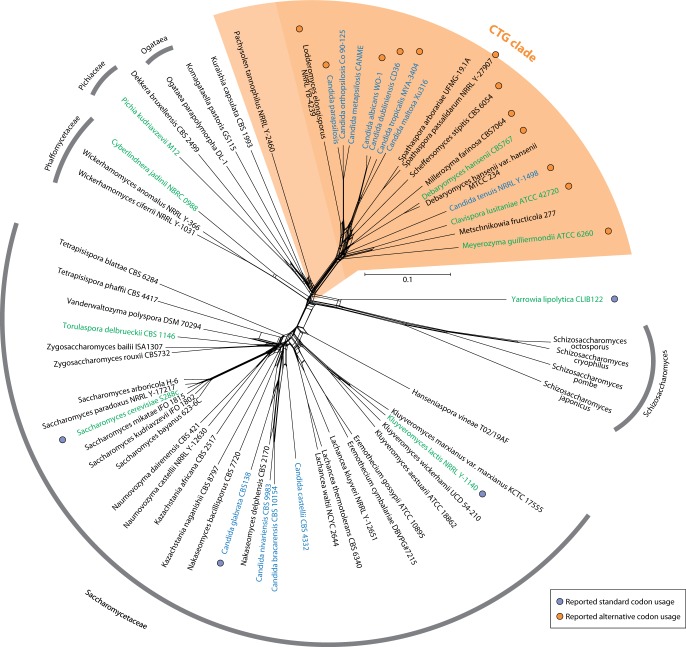

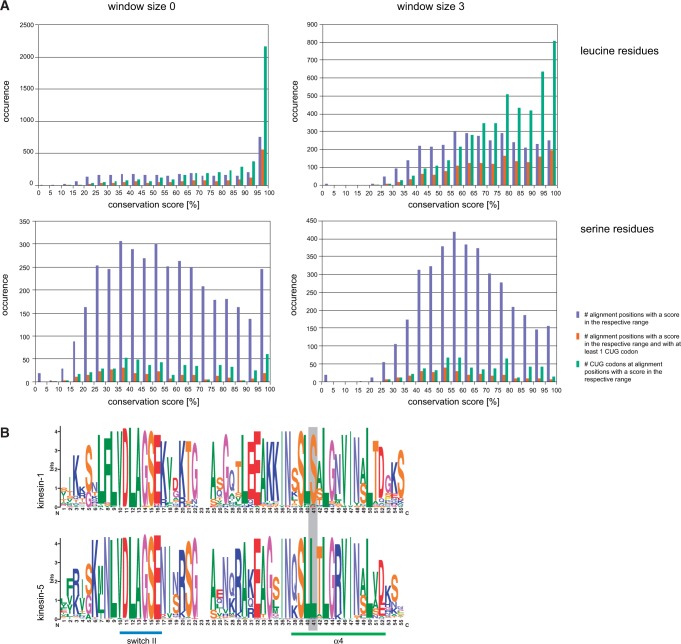
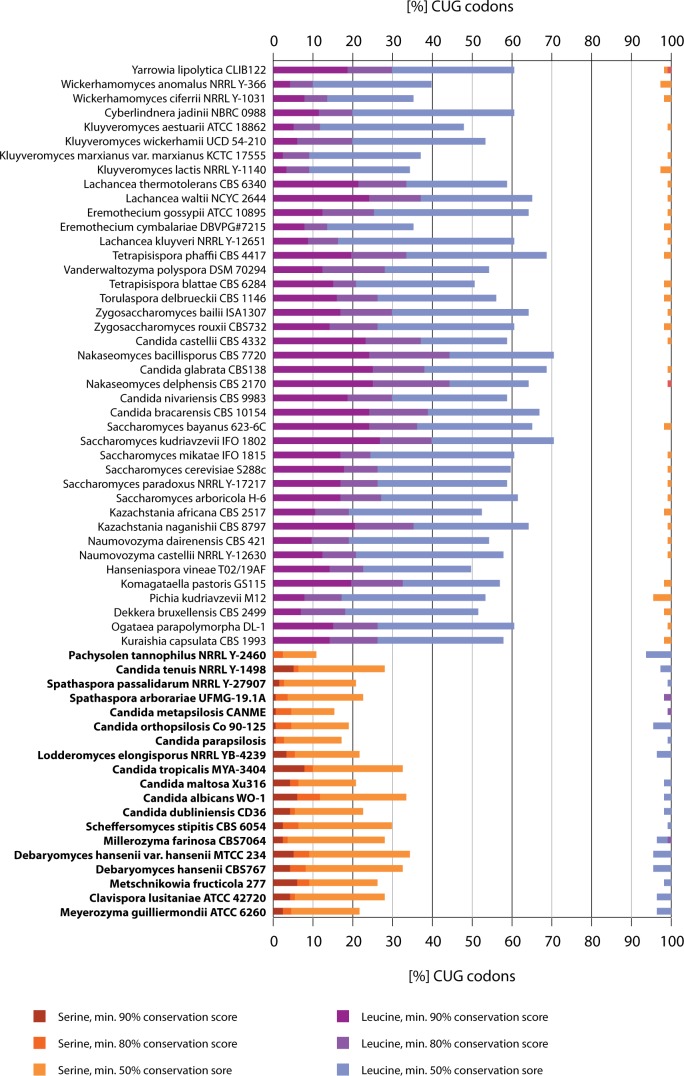
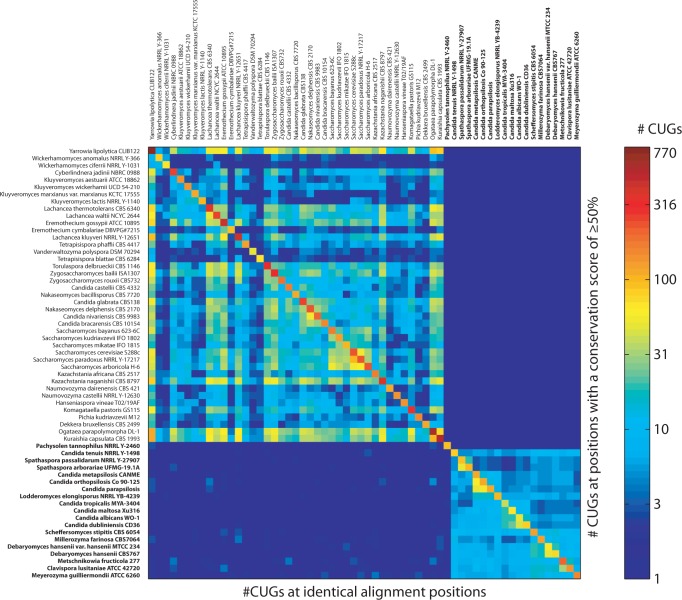
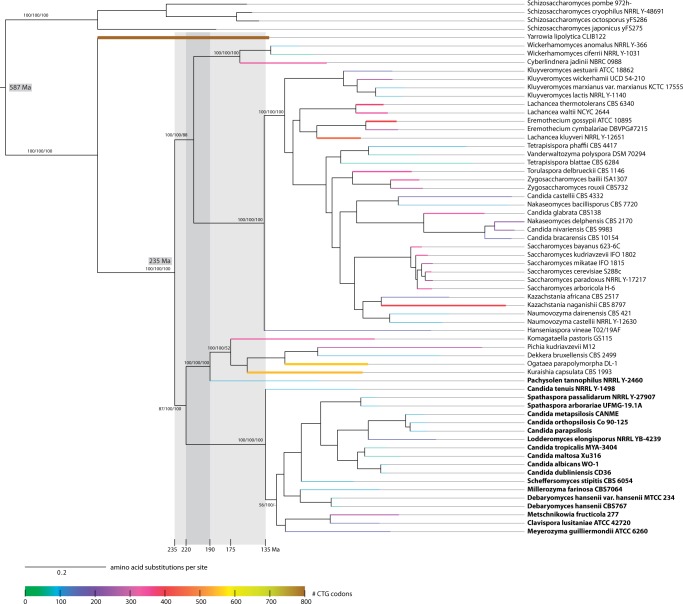
Corrected and republished from
- previous manuscript doi: 10.1093/gbe/evu093
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
