

Bayesian models for syndrome- and gene-specific probabilities of novel variant pathogenicity
- PMID: 25649125
- PMCID: PMC4308924
- DOI: 10.1186/s13073-014-0120-4
Bayesian models for syndrome- and gene-specific probabilities of novel variant pathogenicity
Abstract
Background: With the advent of affordable and comprehensive sequencing technologies, access to molecular genetics for clinical diagnostics and research applications is increasing. However, variant interpretation remains challenging, and tools that close the gap between data generation and data interpretation are urgently required. Here we present a transferable approach to help address the limitations in variant annotation.
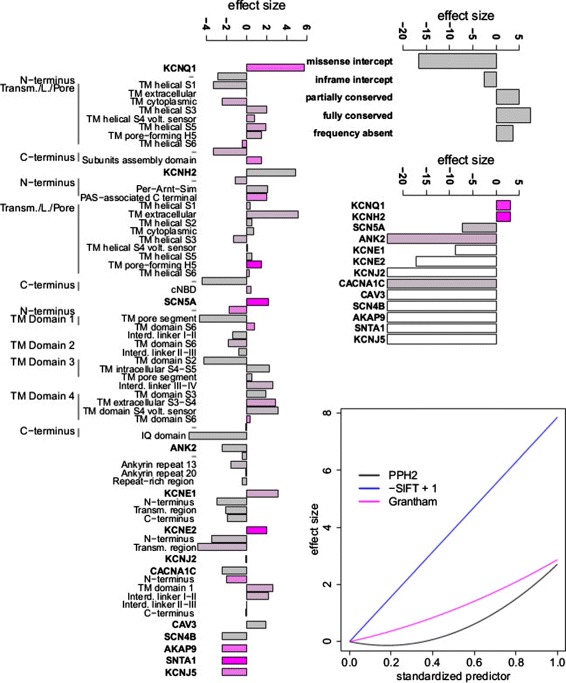
Methods: We develop a network of Bayesian logistic regression models that integrate multiple lines of evidence to evaluate the probability that a rare variant is the cause of an individual's disease. We present models for genes causing inherited cardiac conditions, though the framework is transferable to other genes and syndromes.
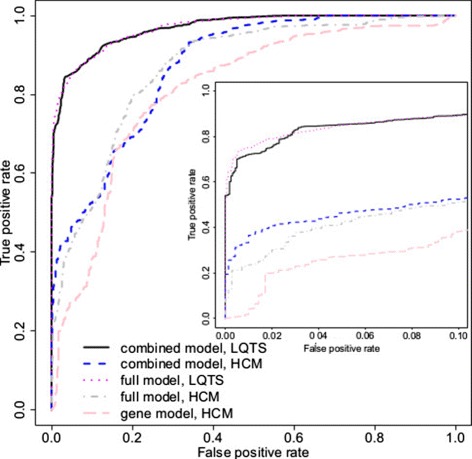
Results: Our models report a probability of pathogenicity, rather than a categorisation into pathogenic or benign, which captures the inherent uncertainty of the prediction. We find that gene- and syndrome-specific models outperform genome-wide approaches, and that the integration of multiple lines of evidence performs better than individual predictors. The models are adaptable to incorporate new lines of evidence, and results can be combined with familial segregation data in a transparent and quantitative manner to further enhance predictions. Though the probability scale is continuous, and innately interpretable, performance summaries based on thresholds are useful for comparisons. Using a threshold probability of pathogenicity of 0.9, we obtain a positive predictive value of 0.999 and sensitivity of 0.76 for the classification of variants known to cause long QT syndrome over the three most important genes, which represents sufficient accuracy to inform clinical decision-making. A web tool APPRAISE [http://www.cardiodb.org/APPRAISE] provides access to these models and predictions.
Conclusions: Our Bayesian framework provides a transparent, flexible and robust framework for the analysis and interpretation of rare genetic variants. Models tailored to specific genes outperform genome-wide approaches, and can be sufficiently accurate to inform clinical decision-making.
Figures
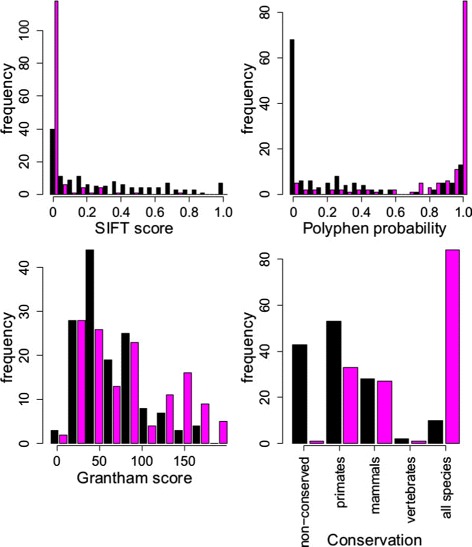
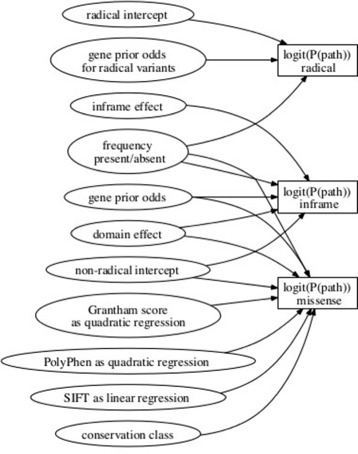
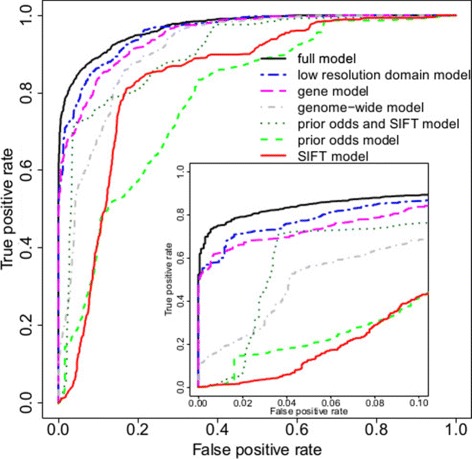
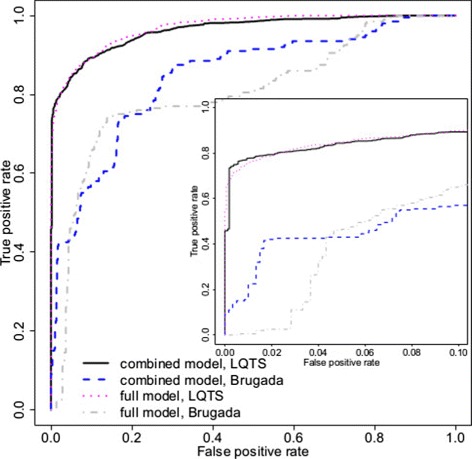
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
