

The 3' addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1
- PMID: 25652405
- PMCID: PMC4406295
- DOI: 10.1093/hmg/ddv044
The 3' addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1
Abstract
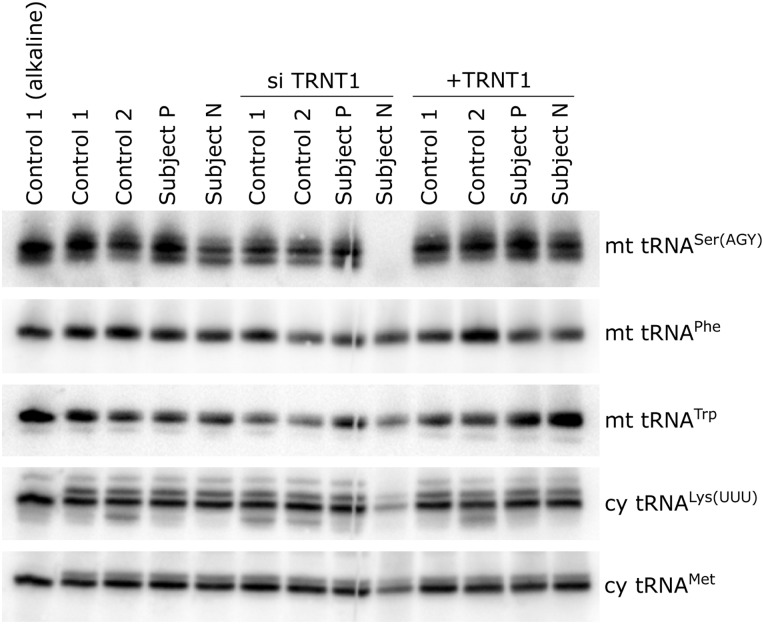
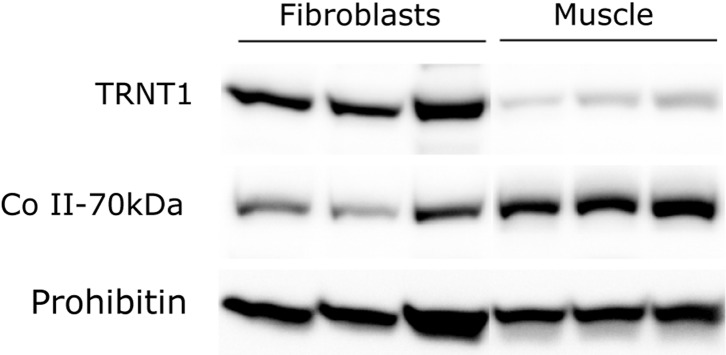
Addition of the trinucleotide cytosine/cytosine/adenine (CCA) to the 3' end of transfer RNAs (tRNAs) is essential for translation and is catalyzed by the enzyme TRNT1 (tRNA nucleotidyl transferase), which functions in both the cytoplasm and mitochondria. Exome sequencing revealed TRNT1 mutations in two unrelated subjects with different clinical features. The first presented with acute lactic acidosis at 3 weeks of age and developed severe developmental delay, hypotonia, microcephaly, seizures, progressive cortical atrophy, neurosensorial deafness, sideroblastic anemia and renal Fanconi syndrome, dying at 21 months. The second presented at 3.5 years with gait ataxia, dysarthria, gross motor regression, hypotonia, ptosis and ophthalmoplegia and had abnormal signals in brainstem and dentate nucleus. In subject 1, muscle biopsy showed combined oxidative phosphorylation (OXPHOS) defects, but there was no OXPHOS deficiency in fibroblasts from either subject, despite a 10-fold-reduction in TRNT1 protein levels in fibroblasts of the first subject. Furthermore, in normal controls, TRNT1 protein levels are 10-fold lower in muscle than in fibroblasts. High resolution northern blots of subject fibroblast RNA suggested incomplete CCA addition to the non-canonical mitochondrial tRNA(Ser(AGY)), but no obvious qualitative differences in other mitochondrial or cytoplasmic tRNAs. Complete knockdown of TRNT1 in patient fibroblasts rendered mitochondrial tRNA(Ser(AGY)) undetectable, and markedly reduced mitochondrial translation, except polypeptides lacking Ser(AGY) codons. These data suggest that the clinical phenotypes associated with TRNT1 mutations are largely due to impaired mitochondrial translation, resulting from defective CCA addition to mitochondrial tRNA(Ser(AGY)), and that the severity of this biochemical phenotype determines the severity and tissue distribution of clinical features.
© The Author 2015. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures



References
-
- Suzuki T., Nagao A., Suzuki T. (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet., 45, 299–329. - PubMed
-
- Ghezzi D., Baruffini E., Haack T.B., Invernizzi F., Melchionda L., Dallabona C., Strom T.M., Parini R., Burlina A.B., Meitinger T., et al. (2012) Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am. J. Hum. Genet., 90, 1079–1087. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
