

Nascent transcription affected by RNA polymerase IV in Zea mays
- PMID: 25653306
- PMCID: PMC4391576
- DOI: 10.1534/genetics.115.174714
Nascent transcription affected by RNA polymerase IV in Zea mays
Abstract
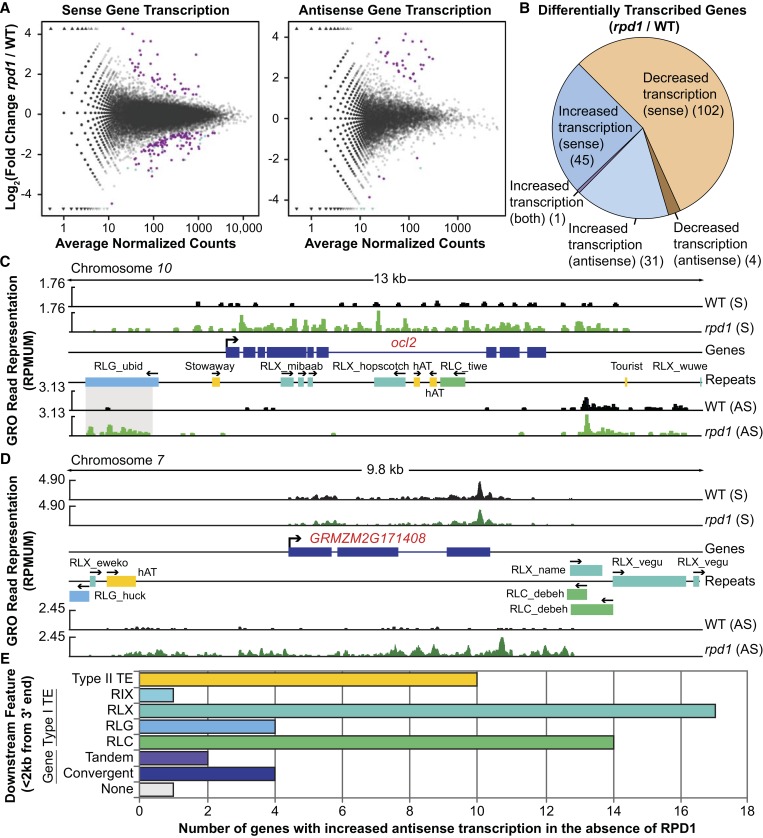
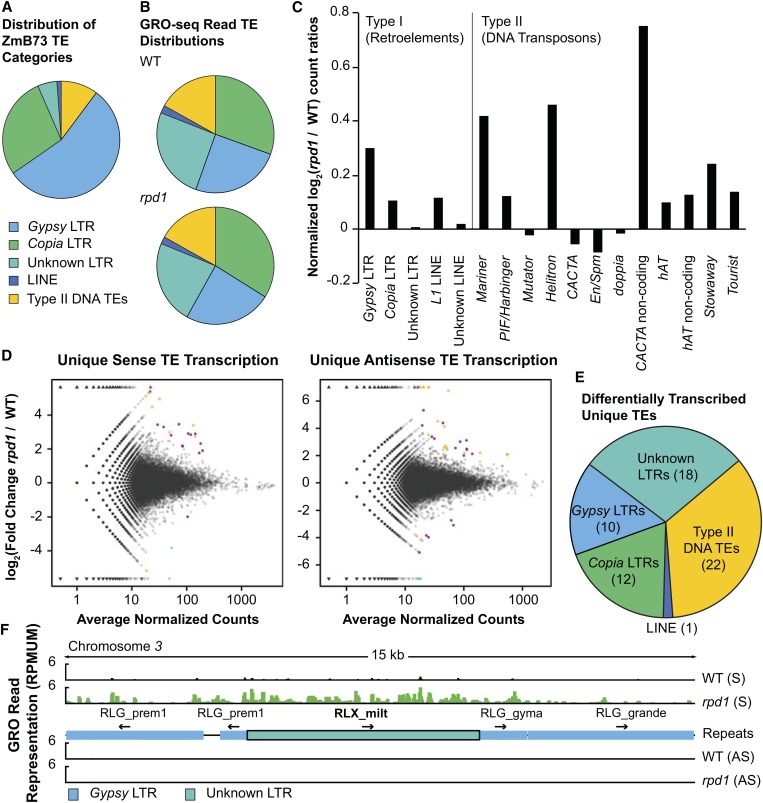
All eukaryotes use three DNA-dependent RNA polymerases (RNAPs) to create cellular RNAs from DNA templates. Plants have additional RNAPs related to Pol II, but their evolutionary role(s) remain largely unknown. Zea mays (maize) RNA polymerase D1 (RPD1), the largest subunit of RNA polymerase IV (Pol IV), is required for normal plant development, paramutation, transcriptional repression of certain transposable elements (TEs), and transcriptional regulation of specific alleles. Here, we define the nascent transcriptomes of rpd1 mutant and wild-type (WT) seedlings using global run-on sequencing (GRO-seq) to identify the broader targets of RPD1-based regulation. Comparisons of WT and rpd1 mutant GRO-seq profiles indicate that Pol IV globally affects transcription at both transcriptional start sites and immediately downstream of polyadenylation addition sites. We found no evidence of divergent transcription from gene promoters as seen in mammalian GRO-seq profiles. Statistical comparisons identify genes and TEs whose transcription is affected by RPD1. Most examples of significant increases in genic antisense transcription appear to be initiated by 3'-proximal long terminal repeat retrotransposons. These results indicate that maize Pol IV specifies Pol II-based transcriptional regulation for specific regions of the maize genome including genes having developmental significance.
Keywords: RNA polymerase IV; gene regulation; paramutation; transcription; transposons.
Copyright © 2015 by the Genetics Society of America.
Figures
References
-
- Alleman M., Sidorenko L., McGinnis K., Seshadri V., Dorweiler J. E., et al. , 2006. An RNA-dependent RNA polymerase is required for paramutation in maize. Nature 442: 295–298. - PubMed
-
- Ariel F., Jegu T., Latrasse D., Romero-Barrios N., Christ A., et al. , 2014. Noncoding transcription by alternative RNA polymerases dynamically regulates an auxin-driven chromatin loop. Mol. Cell 55: 383–396. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
