

Familial lecithin-cholesterol acyltransferase (LCAT) deficiency; a differential of proteinuria
- PMID: 25657982
- PMCID: PMC4316582
- DOI: 10.12860/jnp.2015.05
Familial lecithin-cholesterol acyltransferase (LCAT) deficiency; a differential of proteinuria
Abstract
Background: Lecithin cholesterol acyltransferase (LCAT) is an important enzyme in cholesterol metabolism that is involved in the esterification of cholesterol. A lack of this enzyme results in deranged metabolic pathways that are not completely understood, resulting in abnormal deposition of lipids in several organs. Clinically, it manifests with proteinuria, dyslipidemia and corneal opacity with progressive chronic kidney disease resulting in end-stage renal disease.
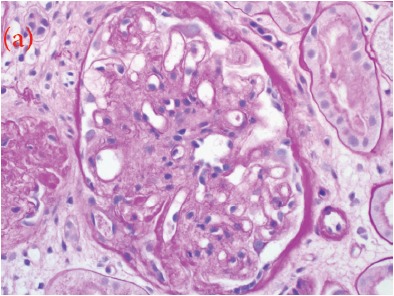
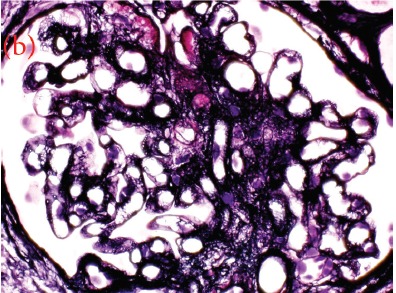
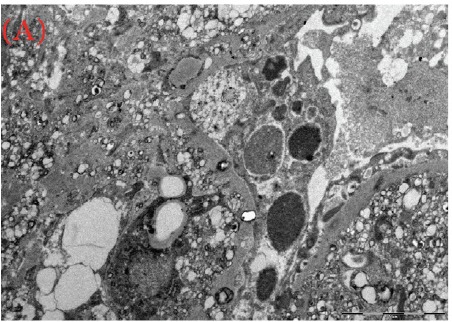
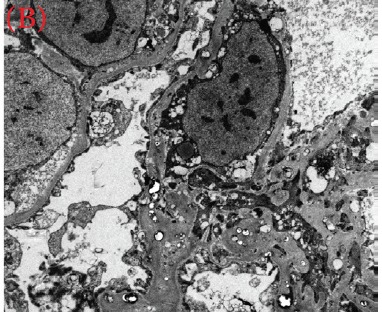
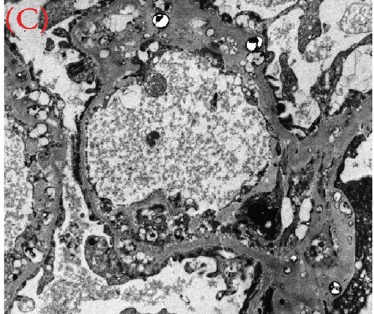
Case presentation: We herein present a case of a 30-year-old male with proteinuria that was not responsive to empiric management with angiotensin-converting enzyme (ACE) inhibitors and oral steroids. Physical examination revealed corneal ring opacity involving both eyes. Urinalysis revealed an active sediment. The 24-h proteinuria was 3.55 grams. Family history was positive for renal disease and dyslipidemia. Viral serology for human immunodeficiency virus (HIV), hepatitis C virus (HCV) and hepatitis B virus (HBV) were negative. Serum complements were normal and anti-nuclear antibody (ANA) was negative. We elected for a renal biopsy that revealed characteristic features of LCAT deficiency. The diagnosis of LCAT deficiency was established with a combination of clinical and pathological findings.
Conclusions: Currently renal prognosis is poor but conservative management with ACE inhibitors and lipid lowering therapy in addition to steroids has been shown to retard progression to end-stage renal disease. However newer therapies such as gene replacement and recombinant LCAT replacement are being studied with promising preliminary results.
Keywords: Familial lecithin-cholesterol acyltransferase (LCAT) deficiency; HDL-cholesterol; Proteinuria.

References
-
- Funke H, von Eckardstein A, Pritchard PH, Albers JJ, Kastelein JJ, Droste C. et al. A molecular defect causing fish eye disease: an amino acid exchange in lecithin-cholesterol acyltransferase (LCAT) leads to the selective loss of alpha-LCAT activity. Proc Natl Acad Sci U S A. 1991;88(11):4855–9. - PMC - PubMed
-
- Klein HG, Lohse P, Pritchard PH, Bojanovski D, Schmidt H, Brewer HB Jr. Two different allelic mutations in the lecithin-cholesterol acyltransferase gene associated with the fish eye syndrome Lecithin-cholesterol acyltransferase (Thr123----Ile) and lecithin-cholesterol acyltransferase (Thr347----Met) J Clin Invest. 1992;89(2):499–506. - PMC - PubMed
-
- Gigante M, Ranieri E, Cerullo G, Calabresi L, Iolascon A, Assmann G. et al. LCAT deficiency: molecular and phenotypic characterization of an Italian family. J Nephrol. 2006;19(3):375–81. - PubMed
-
- Chen C, Applegate K, King WC, Glomset JA, Norum KR, Gjone E. A study of the small spherical high density lipoproteins of patients afflicted with familial lecithin: cholesterol acyltransferase deficiency. J Lipid Res. 1984;25(3):269–82. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous