

COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency
- PMID: 25658047
- PMCID: PMC4320255
- DOI: 10.1016/j.ajhg.2014.12.023
COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency
Abstract
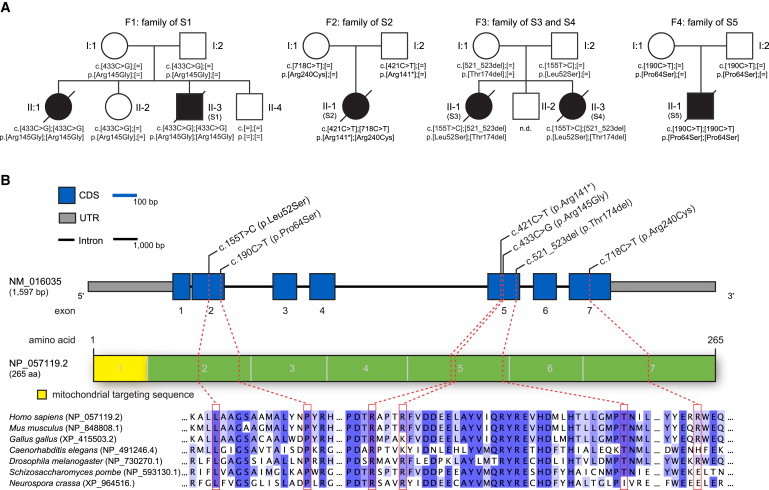
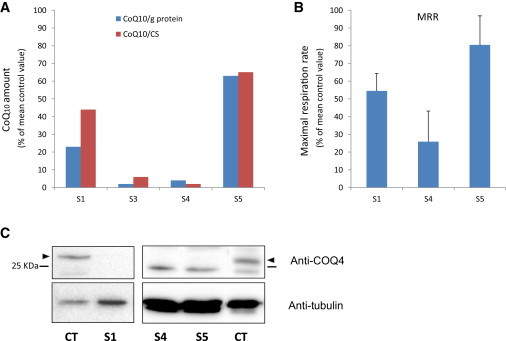
Primary coenzyme Q10 (CoQ10) deficiencies are rare, clinically heterogeneous disorders caused by mutations in several genes encoding proteins involved in CoQ10 biosynthesis. CoQ10 is an essential component of the electron transport chain (ETC), where it shuttles electrons from complex I or II to complex III. By whole-exome sequencing, we identified five individuals carrying biallelic mutations in COQ4. The precise function of human COQ4 is not known, but it seems to play a structural role in stabilizing a multiheteromeric complex that contains most of the CoQ10 biosynthetic enzymes. The clinical phenotypes of the five subjects varied widely, but four had a prenatal or perinatal onset with early fatal outcome. Two unrelated individuals presented with severe hypotonia, bradycardia, respiratory insufficiency, and heart failure; two sisters showed antenatal cerebellar hypoplasia, neonatal respiratory-distress syndrome, and epileptic encephalopathy. The fifth subject had an early-onset but slowly progressive clinical course dominated by neurological deterioration with hardly any involvement of other organs. All available specimens from affected subjects showed reduced amounts of CoQ10 and often displayed a decrease in CoQ10-dependent ETC complex activities. The pathogenic role of all identified mutations was experimentally validated in a recombinant yeast model; oxidative growth, strongly impaired in strains lacking COQ4, was corrected by expression of human wild-type COQ4 cDNA but failed to be corrected by expression of COQ4 cDNAs with any of the mutations identified in affected subjects. COQ4 mutations are responsible for early-onset mitochondrial diseases with heterogeneous clinical presentations and associated with CoQ10 deficiency.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Turunen M., Olsson J., Dallner G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta. 2004;1660:171–199. - PubMed
-
- Echtay K.S., Winkler E., Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature. 2000;408:609–613. - PubMed
-
- Miller R.W., Curry J.R. Mammalian dihydroorotate—ubiquinone reducatse complex. II. Correlation with cytochrome oxidase, mode of linkage with the cytochrome chain, and general properties. Can. J. Biochem. 1969;47:725–734. - PubMed
-
- Schmelzer C., Lorenz G., Rimbach G., Döring F. Influence of Coenzyme Q_10 on release of pro-inflammatory chemokines in the human monocytic cell line THP-1. Biofactors. 2007;31:211–217. - PubMed
-
- Bentinger M., Tekle M., Dallner G. Coenzyme Q—biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010;396:74–79. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
