

Data Imputation in Epistatic MAPs by Network-Guided Matrix Completion
- PMID: 25658751
- PMCID: PMC4449711
- DOI: 10.1089/cmb.2014.0158
Data Imputation in Epistatic MAPs by Network-Guided Matrix Completion
Abstract
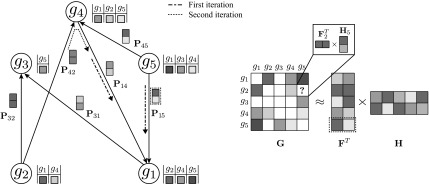
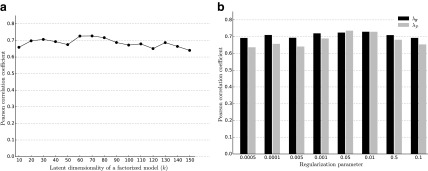
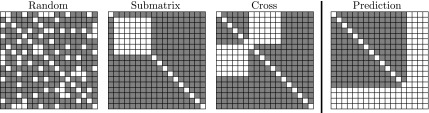
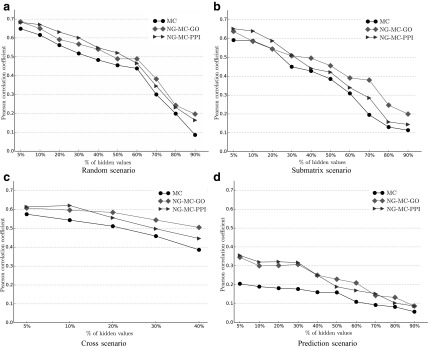
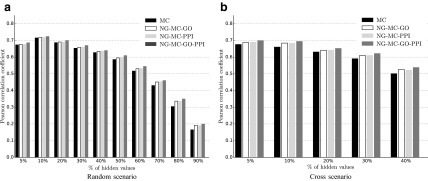
Epistatic miniarray profile (E-MAP) is a popular large-scale genetic interaction discovery platform. E-MAPs benefit from quantitative output, which makes it possible to detect subtle interactions with greater precision. However, due to the limits of biotechnology, E-MAP studies fail to measure genetic interactions for up to 40% of gene pairs in an assay. Missing measurements can be recovered by computational techniques for data imputation, in this way completing the interaction profiles and enabling downstream analysis algorithms that could otherwise be sensitive to missing data values. We introduce a new interaction data imputation method called network-guided matrix completion (NG-MC). The core part of NG-MC is low-rank probabilistic matrix completion that incorporates prior knowledge presented as a collection of gene networks. NG-MC assumes that interactions are transitive, such that latent gene interaction profiles inferred by NG-MC depend on the profiles of their direct neighbors in gene networks. As the NG-MC inference algorithm progresses, it propagates latent interaction profiles through each of the networks and updates gene network weights toward improved prediction. In a study with four different E-MAP data assays and considered protein-protein interaction and gene ontology similarity networks, NG-MC significantly surpassed existing alternative techniques. Inclusion of information from gene networks also allowed NG-MC to predict interactions for genes that were not included in original E-MAP assays, a task that could not be considered by current imputation approaches.
Keywords: data integration; epistatic miniarray profile; gene network; genetic interaction; matrix completion; missing value imputation.
Figures
References
-
- Cai J.-F., Candès E.J., and Shen Z. 2010. A singular value thresholding algorithm for matrix completion. SIAM J. Optimiz. 20, 1956–1982
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
