

Homozygous STIL mutation causes holoprosencephaly and microcephaly in two siblings
- PMID: 25658757
- PMCID: PMC4319975
- DOI: 10.1371/journal.pone.0117418
Homozygous STIL mutation causes holoprosencephaly and microcephaly in two siblings
Abstract
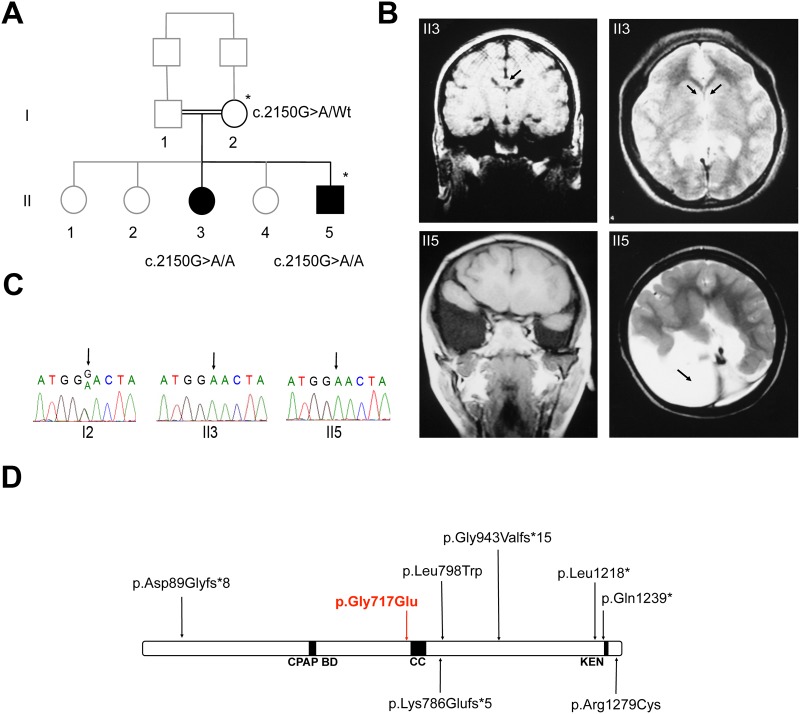
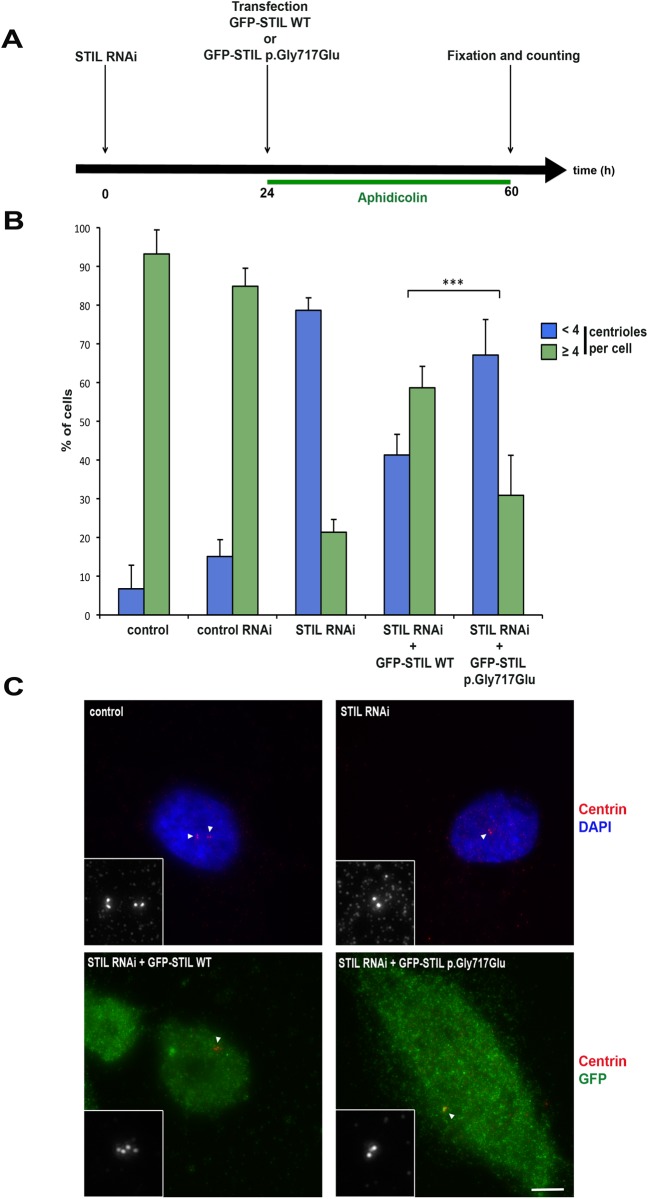
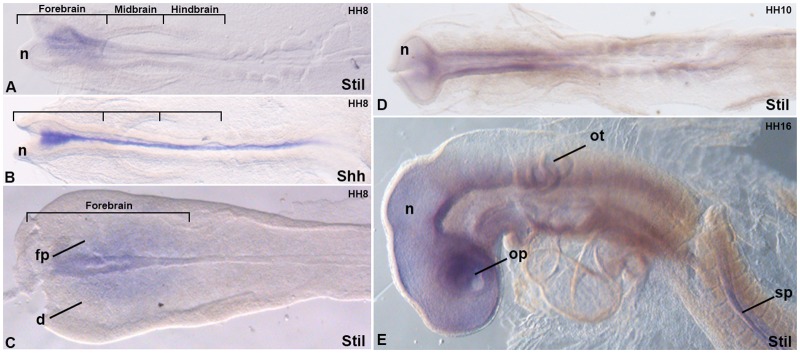
Holoprosencephaly (HPE) is a frequent congenital malformation of the brain characterized by impaired forebrain cleavage and midline facial anomalies. Heterozygous mutations in 14 genes have been identified in HPE patients that account for only 30% of HPE cases, suggesting the existence of other HPE genes. Data from homozygosity mapping and whole-exome sequencing in a consanguineous Turkish family were combined to identify a homozygous missense mutation (c.2150G>A; p.Gly717Glu) in STIL, common to the two affected children. STIL has a role in centriole formation and has previously been described in rare cases of microcephaly. Rescue experiments in U2OS cells showed that the STIL p.Gly717Glu mutation was not able to fully restore the centriole duplication failure following depletion of endogenous STIL protein indicating the deleterious role of the mutation. In situ hybridization experiments using chick embryos demonstrated that expression of Stil was in accordance with a function during early patterning of the forebrain. It is only the second time that a STIL homozygous mutation causing a recessive form of HPE was reported. This result also supports the genetic heterogeneity of HPE and increases the panel of genes to be tested for HPE diagnosis.
Conflict of interest statement
Figures
Similar articles
-
STIL mutation causes autosomal recessive microcephalic lobar holoprosencephaly.Hum Genet. 2015 Jan;134(1):45-51. doi: 10.1007/s00439-014-1487-4. Epub 2014 Sep 14. Hum Genet. 2015. PMID: 25218063
-
Complex mode of inheritance in holoprosencephaly revealed by whole exome sequencing.Clin Genet. 2016 Jun;89(6):659-68. doi: 10.1111/cge.12722. Epub 2016 Feb 16. Clin Genet. 2016. PMID: 26748417
-
A prenatal presentation of severe microcephaly and brain anomalies in a patient with novel compound heterozygous mutations in the STIL gene found postnatally with exome analysis.Pediatr Neurol. 2014 Sep;51(3):434-6. doi: 10.1016/j.pediatrneurol.2014.05.023. Epub 2014 May 29. Pediatr Neurol. 2014. PMID: 24986681
-
Rare nasal cleft in a patient with holoprosencephaly due to a mutation in the ZIC2 gene.Birth Defects Res A Clin Mol Teratol. 2014 Apr;100(4):300-6. doi: 10.1002/bdra.23216. Epub 2014 Feb 12. Birth Defects Res A Clin Mol Teratol. 2014. PMID: 24677696 Review.
-
Holoprosencephaly.Orphanet J Rare Dis. 2007 Feb 2;2:8. doi: 10.1186/1750-1172-2-8. Orphanet J Rare Dis. 2007. PMID: 17274816 Free PMC article. Review.
Cited by
-
Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH).Genet Res (Camb). 2018 Aug 8;100:e7. doi: 10.1017/S0016672318000046. Genet Res (Camb). 2018. PMID: 30086807 Free PMC article. Review.
-
Holoprosencephaly with Clefts: Data of 85 Patients, Treatment and Outcome: Part 1: History, Subdivisions, and Data on 85 Holoprosencephalic Cleft Patients.Ann Maxillofac Surg. 2019 Jan-Jun;9(1):140-145. doi: 10.4103/ams.ams_50_19. Ann Maxillofac Surg. 2019. PMID: 31293943 Free PMC article.
-
Congenital Microcephaly: A Debate on Diagnostic Challenges and Etiological Paradigm of the Shift from Isolated/Non-Syndromic to Syndromic Microcephaly.Cells. 2023 Feb 16;12(4):642. doi: 10.3390/cells12040642. Cells. 2023. PMID: 36831309 Free PMC article. Review.
-
Loss-of-function mutations in FGF8 can be independent risk factors for holoprosencephaly.Hum Mol Genet. 2018 Jun 1;27(11):1989-1998. doi: 10.1093/hmg/ddy106. Hum Mol Genet. 2018. PMID: 29584859 Free PMC article.
-
Whole Exome Sequencing of Thymoma Patients Exhibiting Exceptional Responses to Pemetrexed Monotherapy.Cancers (Basel). 2023 Aug 8;15(16):4018. doi: 10.3390/cancers15164018. Cancers (Basel). 2023. PMID: 37627046 Free PMC article.
References
-
- Solomon BD, Gropman A, Muenke M (1993) Holoprosencephaly Overview. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR et al., editors. GeneReviews(R) Seattle (WA).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
