

Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1-42-induced Alzheimer's disease model rats
- PMID: 25658940
- PMCID: PMC4319949
- DOI: 10.1371/journal.pone.0116549
Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1-42-induced Alzheimer's disease model rats
Abstract
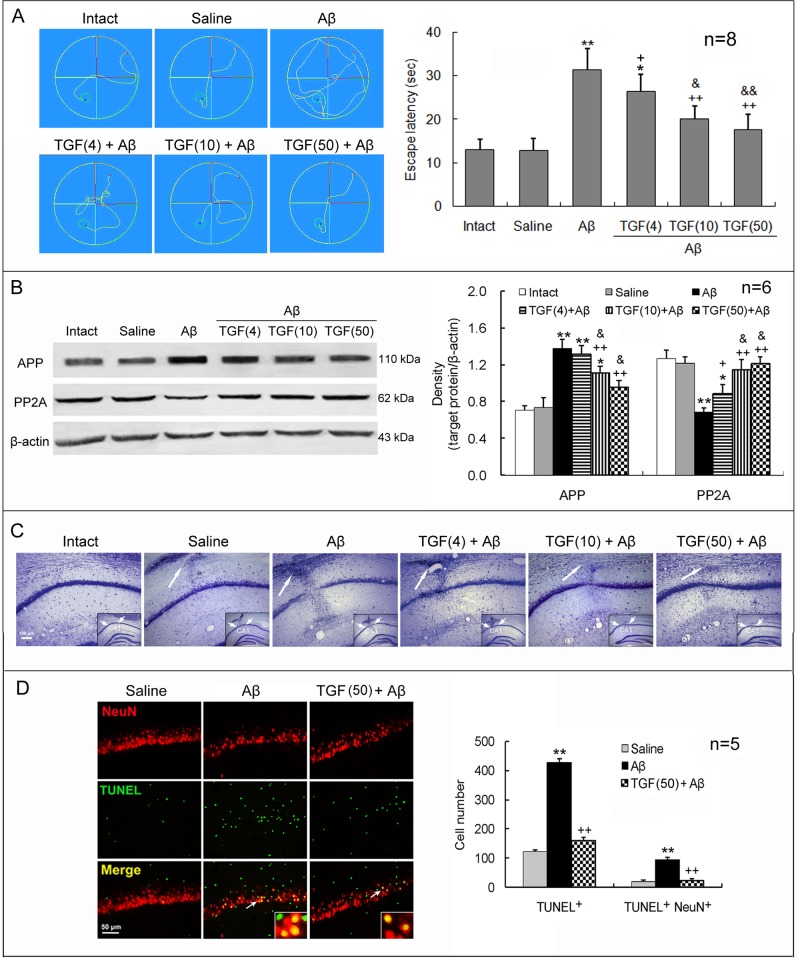
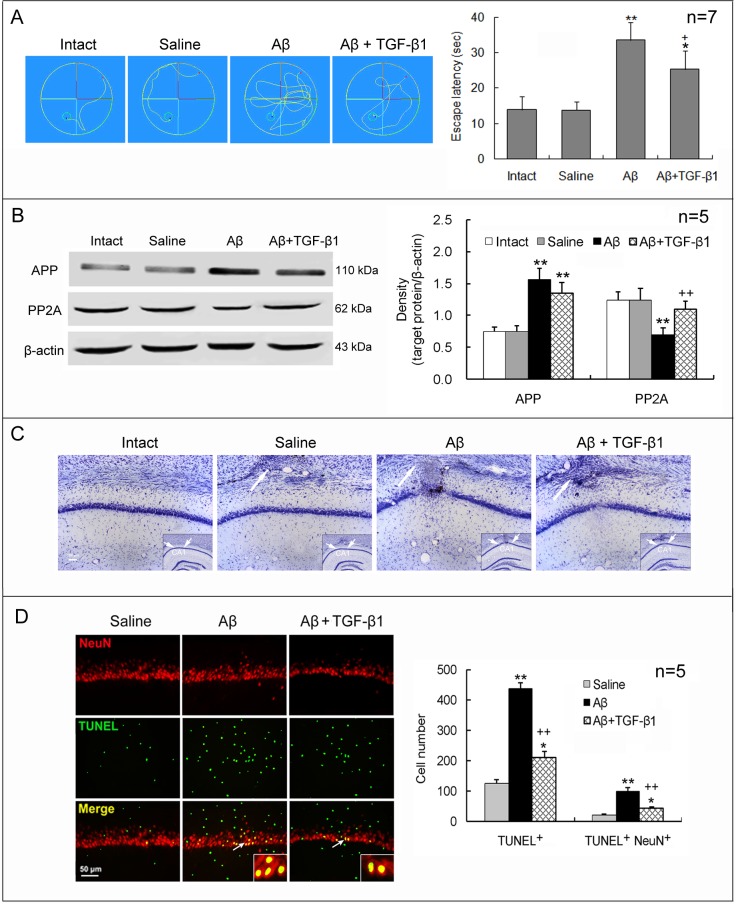
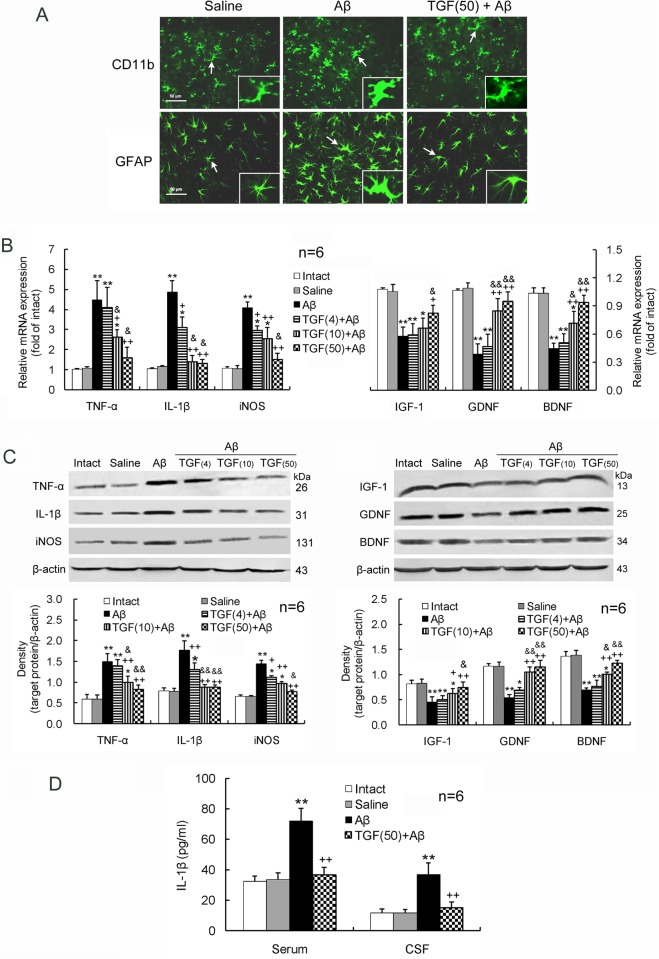
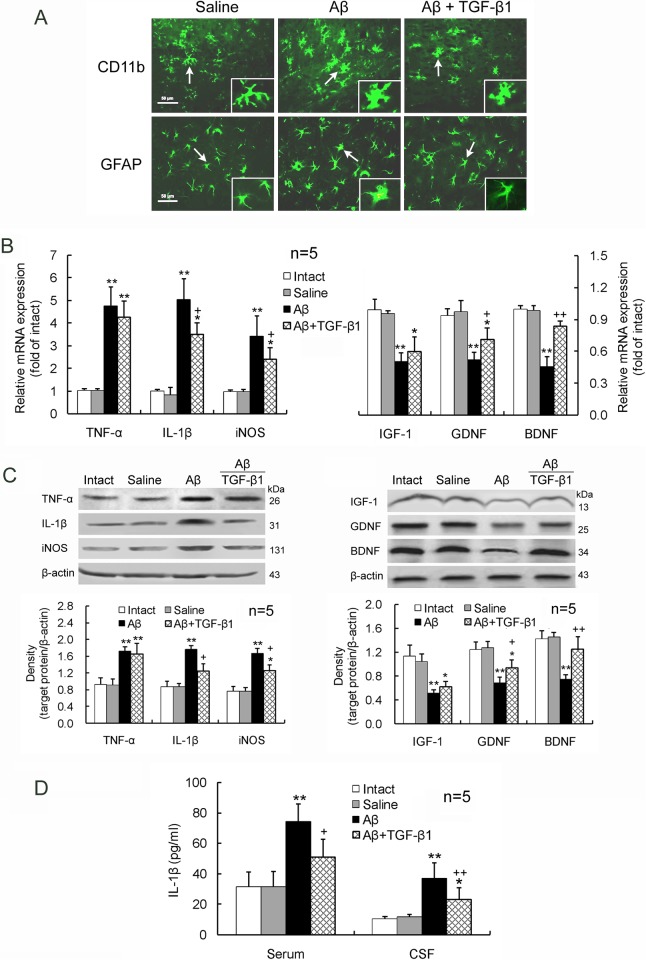
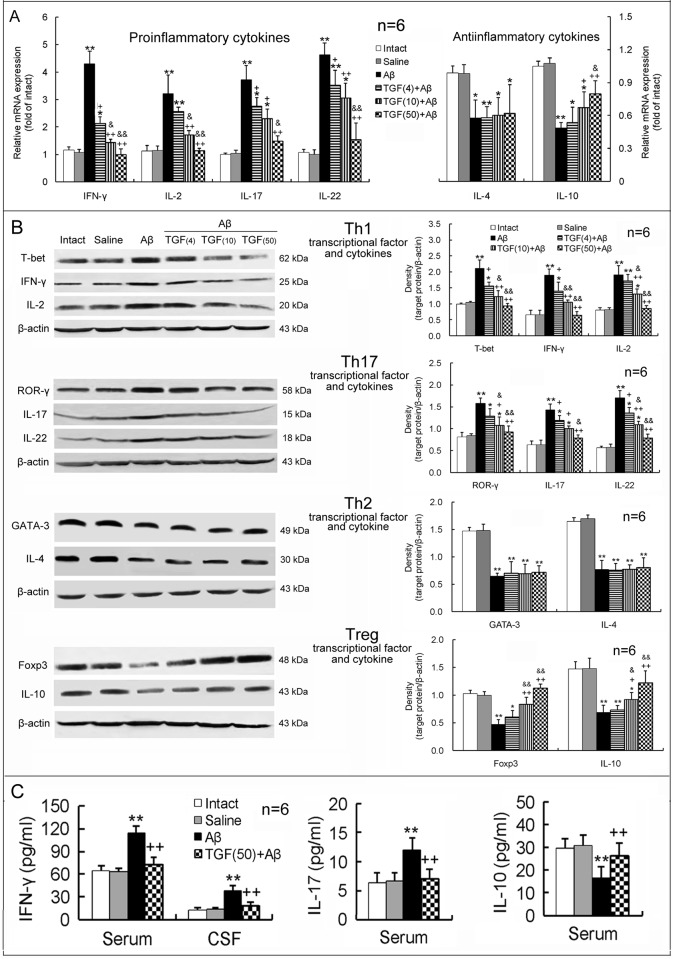
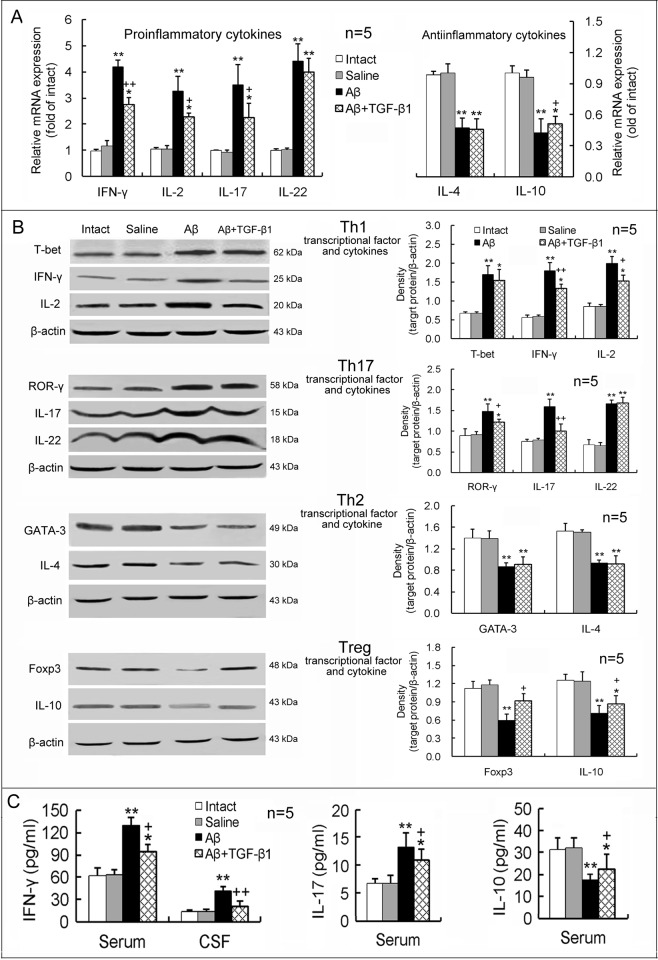
Neuroinflammation has been reported to be associated with Alzheimer's disease (AD) pathogenesis. Neuroinflammation is generally considered as an outcome of glial activation; however, we recently demonstrated that T helper (Th)17 cells, a subpopulation of proinflammatory CD4+ T cells, are also involved in AD pathogenesis. Transforming growth factor (TGF)-β1, a cytokine that can be expressed in the brain, can be immunosuppressive, but its effects on lymphocyte-mediated neuroinflammation in AD pathogenesis have not been well addressed. In the current study we administered TGF-β1 via intracerebroventricle (ICV) and intranasal (IN) routes in AD model rats to investigate its antiinflammatory and neuroprotective effects. The AD rat model was prepared by bilateral hippocampal injection of amyloid-β (Aβ)1-42. TGF-β1 was administered via ICV one hour prior to Aβ1-42 injection or via both nares seven days after Aβ1-42 injection. ICV administration of TGF-β1 before Aβ1-42 injection remarkably ameliorated Aβ1-42-induced neurodegeneration and prevented Aβ1-42-induced increases in glia-derived proinflammatory mediators (TNF-α, IL-1β and iNOS), as well as T cell-derived proinflammatory cytokines (IFN-γ, IL-2, IL-17 and IL-22), in the hypothalamus, serum or cerebrospinal fluid (CSF) in a concentration-dependent manner. TGF-β1 pretreatment also prevented Aβ1-42-induced decreases in the neurotrophic factors, IGF-1, GDNF and BDNF, and in the antiinflammatory cytokine, IL-10. Similarly, IN administration of TGF-β1 after Aβ1-42 injection reduced neurodegeneration, elevation of proinflammatory mediators and cytokines, and reduction of neurotrophic and antiinflammatory factors, in the hypothalamus, serum or CSF. These findings suggest that TGF-β1 suppresses glial and T cell-mediated neuroinflammation and thereby alleviates AD-related neurodegeneration. The effectiveness of IN administered TGF-β1 in reducing Aβ1-42 neurotoxicity suggests a possible therapeutic approach in patients with AD.
Conflict of interest statement
Figures
References
-
- Schwab C, McGeer PL (2008) Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J Alzheimers Dis 13: 359–369. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
