

MIPSTR: a method for multiplex genotyping of germline and somatic STR variation across many individuals
- PMID: 25659649
- PMCID: PMC4417122
- DOI: 10.1101/gr.182212.114
MIPSTR: a method for multiplex genotyping of germline and somatic STR variation across many individuals
Erratum in
-
Corrigendum: MIPSTR: a method for multiplex genotyping of germline and somatic STR variation across many individuals.Genome Res. 2015 Aug;25(8):1244. Genome Res. 2015. PMID: 26240161 Free PMC article. No abstract available.
Abstract
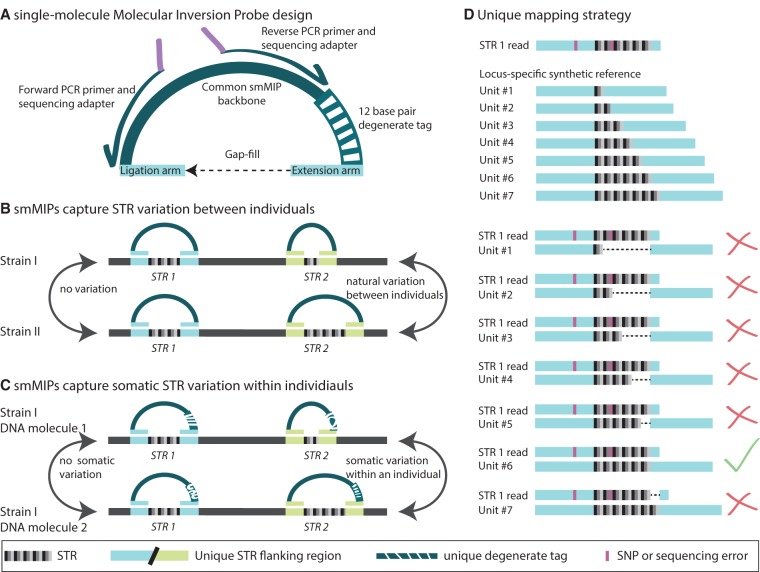
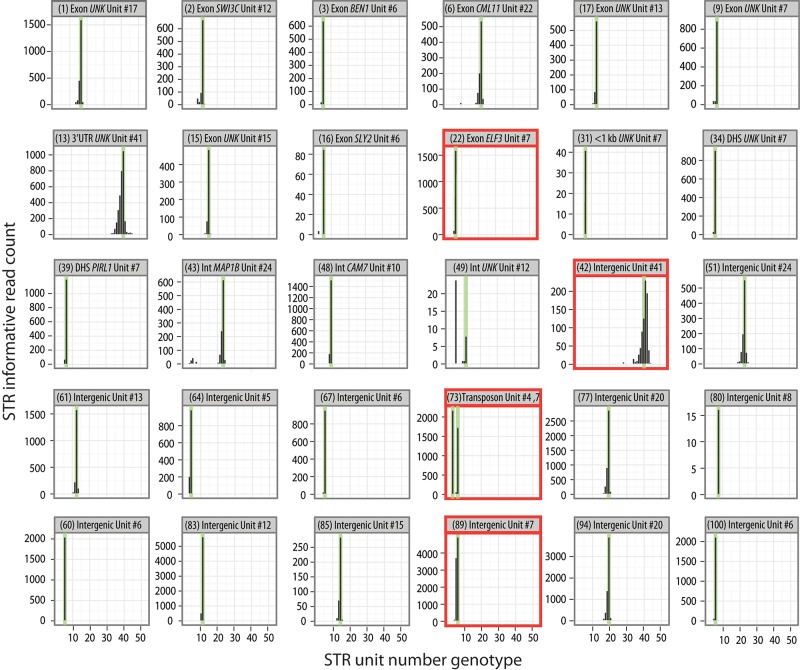
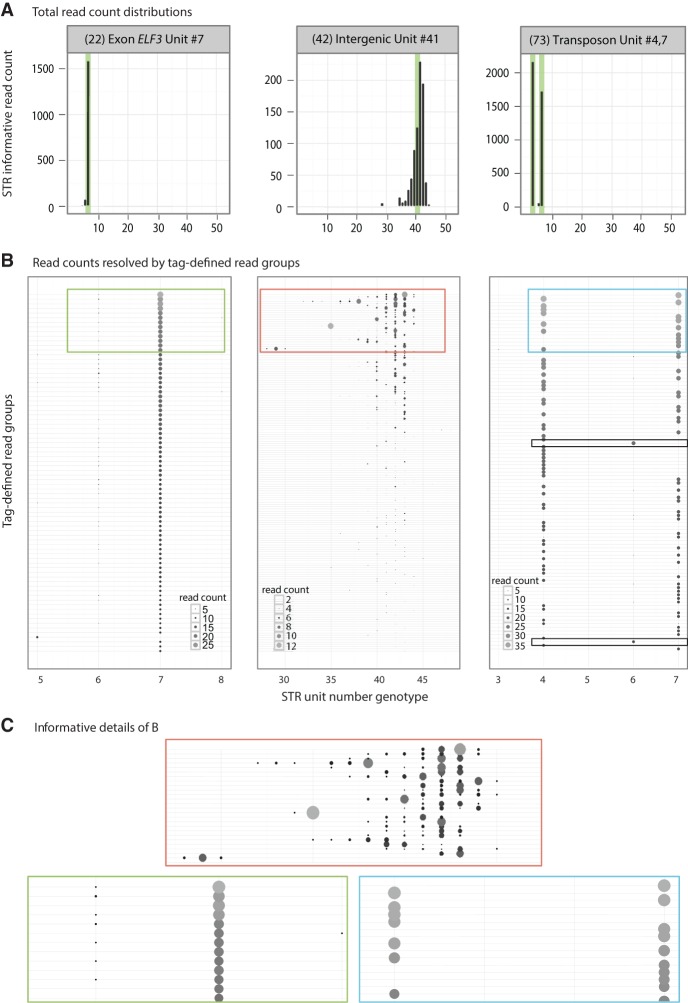
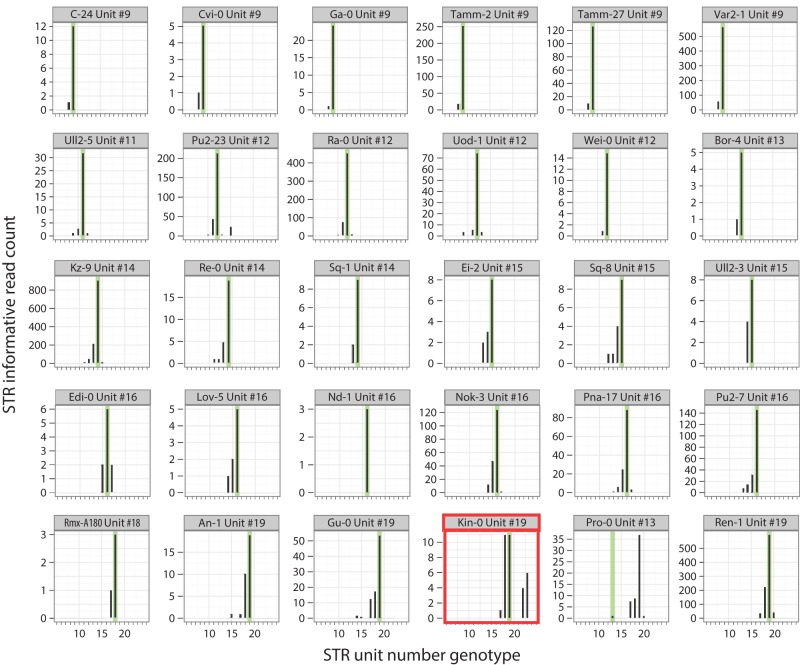
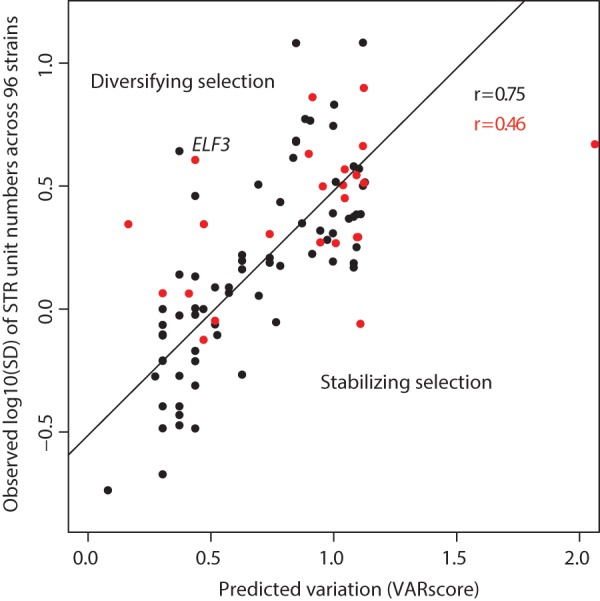
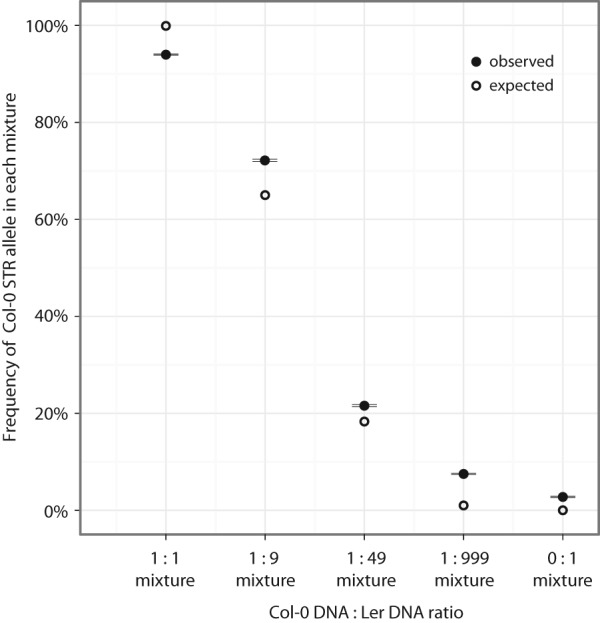
Short tandem repeats (STRs) are highly mutable genetic elements that often reside in regulatory and coding DNA. The cumulative evidence of genetic studies on individual STRs suggests that STR variation profoundly affects phenotype and contributes to trait heritability. Despite recent advances in sequencing technology, STR variation has remained largely inaccessible across many individuals compared to single nucleotide variation or copy number variation. STR genotyping with short-read sequence data is confounded by (1) the difficulty of uniquely mapping short, low-complexity reads; and (2) the high rate of STR amplification stutter. Here, we present MIPSTR, a robust, scalable, and affordable method that addresses these challenges. MIPSTR uses targeted capture of STR loci by single-molecule Molecular Inversion Probes (smMIPs) and a unique mapping strategy. Targeted capture and our mapping strategy resolve the first challenge; the use of single molecule information resolves the second challenge. Unlike previous methods, MIPSTR is capable of distinguishing technical error due to amplification stutter from somatic STR mutations. In proof-of-principle experiments, we use MIPSTR to determine germline STR genotypes for 102 STR loci with high accuracy across diverse populations of the plant A. thaliana. We show that putatively functional STRs may be identified by deviation from predicted STR variation and by association with quantitative phenotypes. Using DNA mixing experiments and a mutant deficient in DNA repair, we demonstrate that MIPSTR can detect low-frequency somatic STR variants. MIPSTR is applicable to any organism with a high-quality reference genome and is scalable to genotyping many thousands of STR loci in thousands of individuals.
© 2015 Carlson et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al.1998. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58: 5248–5257. - PubMed
-
- Butler AP, Trono D, Coletta LD, Beard R, Fraijo R, Kazianis S, Nairn RS. 2007. Regulation of CDKN2A/B and Retinoblastoma genes in Xiphophorus melanoma. Comp Biochem Physiol C Toxicol Pharmacol 145: 145–155. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources