

Review
doi: 10.1021/cr500279h.
Epub 2015 Feb 9.
Electrochemistry of nonconjugated proteins and glycoproteins. Toward sensors for biomedicine and glycomics
Affiliations
- PMID: 25659975
- PMCID: PMC4360380
- DOI: 10.1021/cr500279h
Item in Clipboard
Review
Electrochemistry of nonconjugated proteins and glycoproteins. Toward sensors for biomedicine and glycomics
Chem Rev.
.
No abstract available
Figures
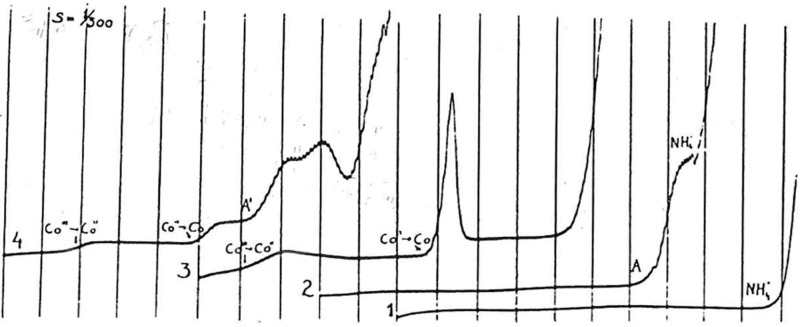
Polarographic
catalytic waves of human serum. (1) Pure supporting
electrolyte, 0.1 M ammonia/ammonium chloride buffer; (2) the “presodium”
catalytic wave, 400-times diluted human serum (A) in 0.1 M ammonia/ammonium
chloride; (3) two-step reduction of Co(III), 1 mM Co(NH3)6Cl3 in 0.1 M ammonia/ammonium chloride; (4)
the catalytic double-wave in Brdička solution, 1 mM Co(NH3)6Cl3 + 400-times diluted human serum
(A′) in 0.1 M ammonia/ammonium chloride; recorded from 0 V
vs mercury pool, 200 mV/abscissa. Adapted with permission from ref (64). Copyright 1933 Collection
of Czechoslovak Chemical Communications.
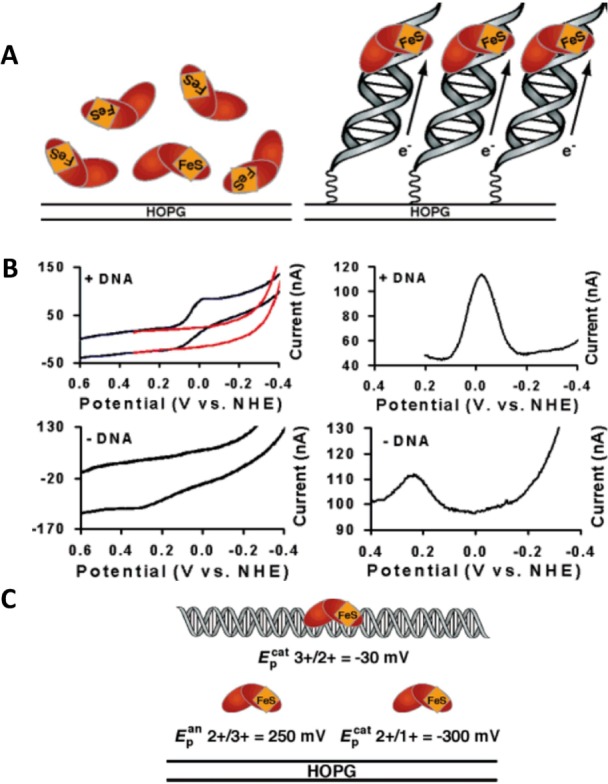
(A) Schematic representation
of electrochemistry for endonuclease
III (Endo III) on highly oriented pyrolytic graphite electrode (HOPGE)
with and without modification with DNA. (B) Cyclic (left, 50 mV/s
scan rate) and square wave voltammograms (right, 15 Hz) of 50 μM
Endo III in 20 mM Na phosphate, 100 mM NaCl, 1 mM EDTA, 20% glycerol,
pH 7.5. The top two panels show electrochemical responses of Endo
III at a HOPGE modified with the sequence pyrene-(CH2)4-Pi-5′-AGT ACA GTC ATC GCG-3′
plus complement. Cyclic voltammograms of a HOPGE modified with DNA
featuring an abasic site are in red (top left), where the abasic position
corresponds to the complement of the italicized base. The bottom two
panels show electrochemical responses of Endo III on a bare HOPGE.
All runs were taken using the inverted drop cell electrode configuration
vs Ag/AgCl reference and Pt auxiliary electrode. (C) Illustration
of the potentials vs normal hydrogen electrode (NHE) of the couples
of Endo III in the presence and absence of DNA. These values are obtained
from SWV on a HOPGE and are averages of at least four trials each.
Adapted with permission from ref (143). Copyright 2006 American Chemical Society.
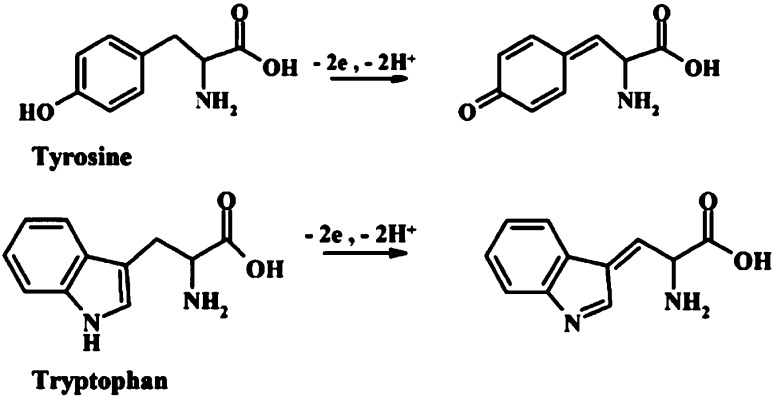
Schemes of EC oxidation
of tyrosine (Tyr) and tryptophan (Trp).
Adapted with permission from ref (164). Copyright 2013 Wiley-VCH Verlag GmbH&Co.
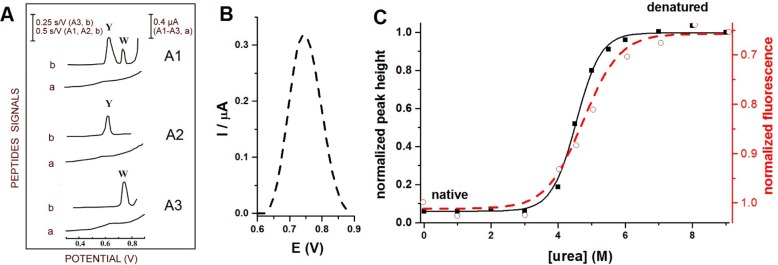
(A) Oxidation of Trp-
and Tyr-containing peptides on carbon paste
electrodes. (a) Differential pulse voltammograms and (b) chronopotentiograms
for (A1) Tyr- and Trp-containing luteinizing hormone releasing hormone,
(A2) Tyr-containing neurotensin, and (A3) Trp-containing bombesin.
10 nM peptide was adsorbed for 5 min at accumulation potential of
0.1 V followed by chronopotentiogram or DP voltammogram recording.
CPS: Istr 5 μA; DPV scan rate, 5
mV/s. Y refers to Tyr and W to Trp residues. Adapted with permission
from ref (159). Copyright
1996 Elsevier. (B) Oxidation peak of 2 μM human serum albumin
(HSA) denatured in 8 M urea at glassy carbon electrode. (C) Dependences
of square wave voltametric peak heights (−■−)
and changes in fluorescence emission at 334 nm (− –○– −)
on urea concentration. 1 μM HSA was incubated overnight with
different urea concentrations (indicated in the figure) at 4 °C.
Oxidation peak height of HSA denatured in 8 M urea was taken as 1.
In the fluorescence measurements, intensity at 334 nm produced by
1 μM HSA incubated in the absence of urea was taken as 1. Adapted
with permission from ref (218). Copyright 2012 Elsevier.
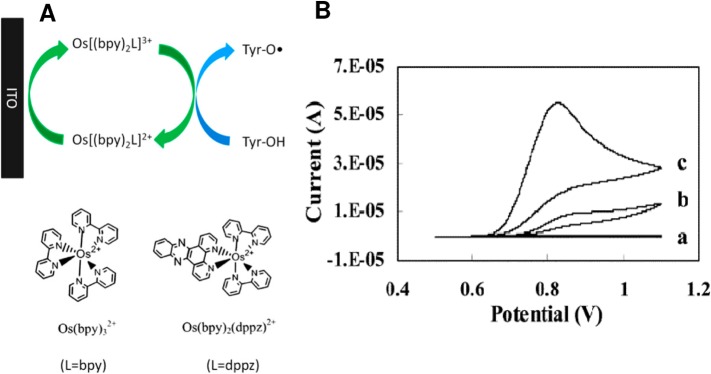
(A) Oxidation of tyrosine (Tyr) with signal enhancement.
ITO, indium
tin oxide electrode; Bipy, 2,2′-bipyridine; dppz, dipyrido
[3,2-a:20,30-c] phenazine. (B) Cyclic
voltammograms of (a) 5 μM Os(bpy)2dppz, (b) 2 mM
Tyr, and (c) 5 μM Os(bpy)2dppz and 2 mM Tyr. Working
electrode: ITO. Reference: Ag/AgCl/3 M KCl. Supporting electrolyte:
100 mM sodium phosphate, pH 7.3. Scan rate: 30 mV/s. Adapted with
permission from ref (185) Copyright 2012 Elsevier.
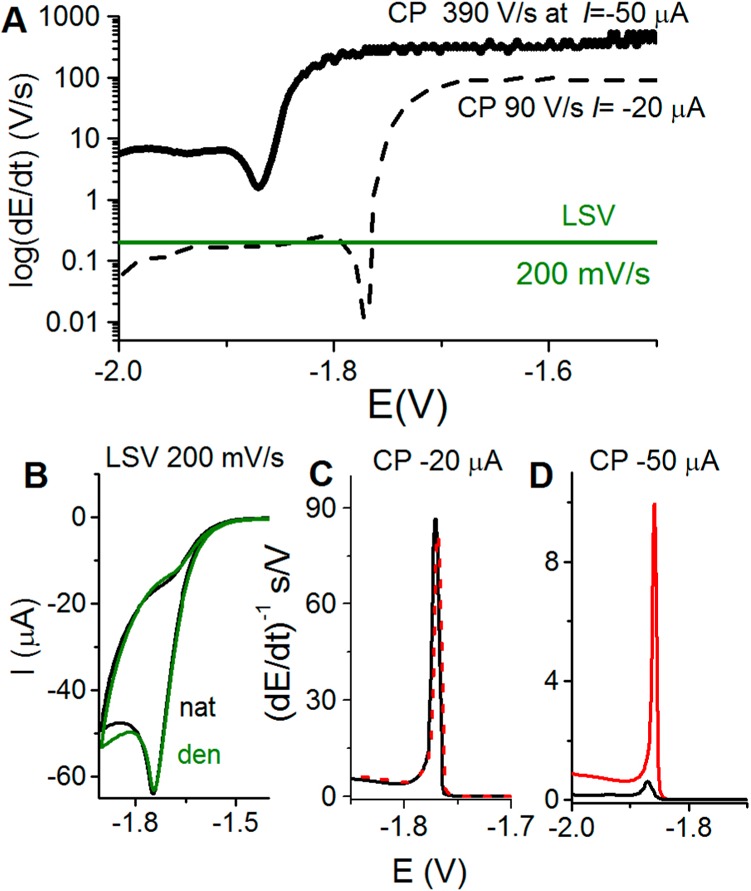
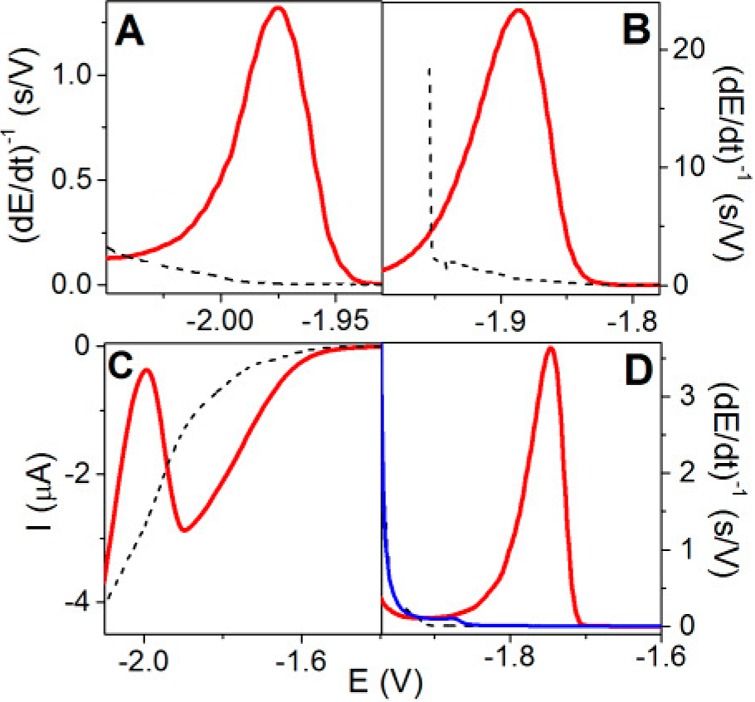
(A) Schematic representation
of the rate of potential changes in
chronopotentiometry (CP) at two different intensities as compared
to voltammetry. In linear sweep voltammetry (LSV), the scan rate (chosen
by an experimenter) is constant throughout the whole voltammogram
recording. However, in CP the rate of potential changes is influenced
by the current density. At constant electrode size, this density is
determined by polarizing current intensity (I, chosen
by an experimenter). In the absence of the electrode process, the
potential changes very rapidly (e.g., 390 V/s at I = −50 μA), but it gets much slower in a narrow potential
range where the electrode process (e.g., proton reduction and hydrogen
evolution) is taking place. (B–D) In protein analysis, both
native (nat, black) and denatured (den, green or red) proteins are
firmly attached to the Hg electrode surface, and prolonged exposure
of native folded protein to negative potentials (at low scan rates
or I intensities) may result in its denaturation,
indicated by almost the same (B) LSV or (C) CP responses. (D) At high
current intensity in CP, for example, at I = −50
μA, fast potential changes (390 V/s) prevent protein from the
denaturation at the negatively charged electrode surface, as indicated
by a relatively small CP response of native (black) protein and a
very large response of the denatured (red) protein.
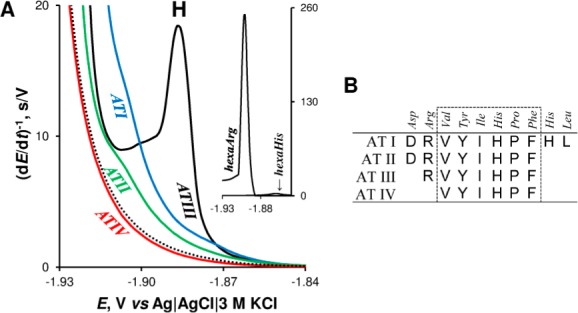
(A)
CPS peaks H of 1 μM angiotensin peptides (AT) in McIlvaine
buffer, pH 7 at HMDE; dotted line represents blank background electrolyte.
Inset: CPS peak H of hexaArg and hexaHis. (B) Amino acids sequences
of AT peptides. Adapted with permission from ref (170). Copyright 2013 Elsevier.
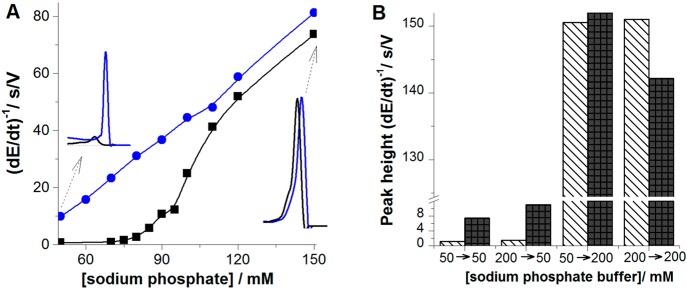
(A) Dependence of peak
height of 100 nM native (black) and denatured
(blue) BSA on concentration of sodium phosphate, pH 7 in the presence
of 56 mM urea (black). Accumulation time tA of 60 s, accumulation potential EA of
−0.1 V, stirring 1500 rpm, stripping current, Istr, of −30 μA. (B) Column graph showing
peak H heights of native (stripped column) and denatured (black column)
BSA obtained in AdT (ex situ) stripping experiment. 100 nM BSA was
adsorbed at HMDE for tA of 60 s at EA of −0.1 V either from 50 mM or from
200 mM sodium phosphate, pH 7, and the BSA-modified electrode was
transferred to the electrolytic cell with blank 50 or 200 mM sodium
phosphate, pH 7, to record the chronopotentiogram. 50 → 200
indicates BSA adsorption from 50 mM phosphate, followed by a transfer
of BSA-modified electrode to 200 mM phosphate in the electrolytic
cell. Denaturation of 14.4 μM BSA in 0.1 M Tris-HCl, pH 7.3,
with 8 M urea was performed overnight at 4 °C. The protein solution
was then diluted by the background electrolyte to the final protein
concentration (usually about 100 nM and immediately measured). Reprinted
with permission from ref (232). Copyright 2009 Royal Society of Chemistry.
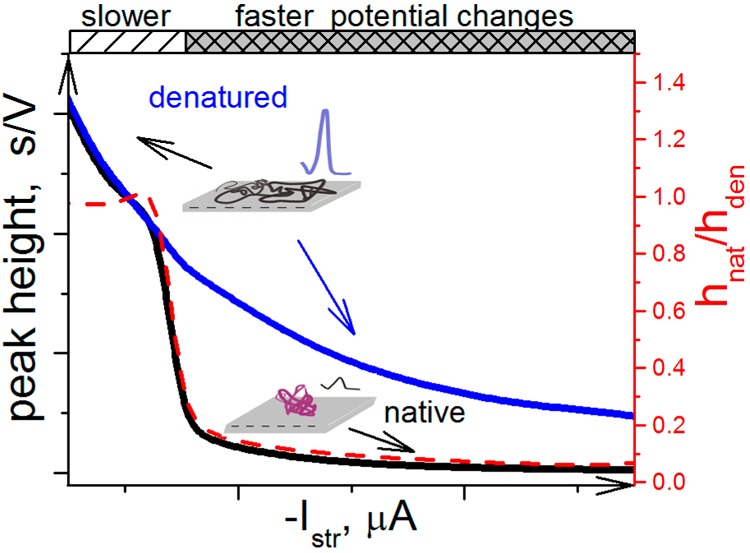
Schematic
representation of the effect of the stripping current
intensity (Istr) on peaks H of native
and denatured proteins. The scheme demonstrates that at low Istr intensities, the surface-attached protein
is denatured, producing almost the same peak H as the protein, which
was denatured in solution by a chemical denaturation agent. The protein
denaturation at the electrode surface is due to the prolonged effect
of the electric field at negative potentials. At higher Istr intensities, the time of exposure of the protein to
negative potentials is much shorter, causing a little harm to the
surface-attached protein as manifested by a relatively small peak
H.
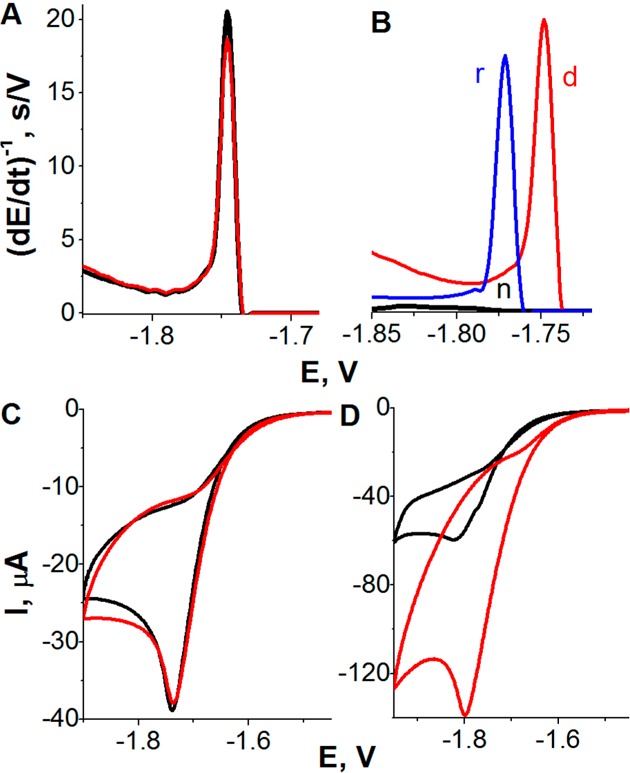
Constant current chronopotentiometric
stripping (CPS) peak H of
100 nM native BSA (n, black), DTT-reduced BSA (r, blue), and guanidinium
chloride (GdmCl)-denatured BSA (d, red) (A) at bare and (B) at DTT-modified
HMDE (DTT-HMDE) in McIlvaine buffer, pH 7. GdmCl was present in all
samples at nondenaturing (70 mM) concentration; conventional CPS measurements
using stripping current, Istr, −70
μA. (C, D) Adsorptive transfer stripping cyclic voltammograms
of native (black) and denatured BSA (red) at DTT-modified HMDE at
scan rates of (C) 50 mV/s and (D) 1 V/s. In this experiment, 1 μM
BSA was adsorbed at DTT-HMDE from McIlvaine buffer, pH 7, for tA 60 s. The adsorptive transfer procedure was
applied to prevent additional BSA adsorption during the potential
scanning. BSA was denatured with 6 M GdmCl. Adapted with permission
from ref (239). Copyright
2010 American Chemical Society.
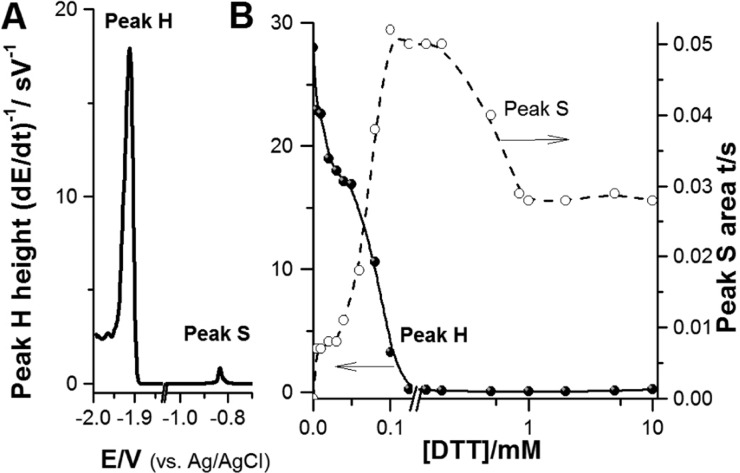
(A) Chronopotentiograms
of 100 nM native BSA coadsorbed with 60
μM DTT at HMDE. (B) Dependence of peak H and peak S heights
obtained with BSA·DTT-HMDE on concentration of DTT. BSA·DTT-HMDE
was prepared by coadsorption of 100 nM BSA and 1 mM DTT at HMDE followed
by CPS peak H recording at stripping current, Istr, −70 μA. Adapted with permission from ref (228). Copyright 2013 Elsevier.
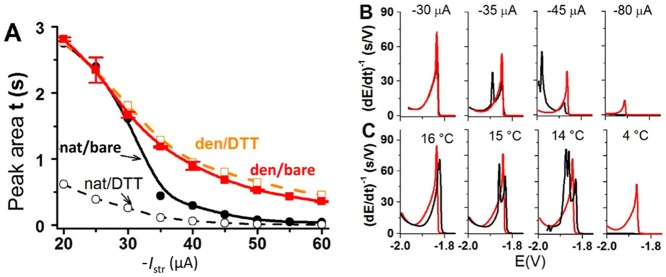
CPS responses of the native and denatured proteins at bare and
DTT-modified mercury electrodes. (A) Dependence of CPS peak H area
of 20 nM native (−●–, − –○– −)
and denatured (−■–, − –□– −)
urease on stripping current, Istr, at
bare (solid line) and DTT-modified (dashed) HMDEs. Urease was adsorbed
at accumulation potential of −0.1 V for accumulation time, tA, of 60 s from McIlvaine buffer, pH 7, with
26 mM GdmCl in a thermostated electrolytic cell at 25 °C, and
CPS analysis proceeded at the given Istr. (B,C) Chronopotentiograms of 20 nM native (black) and denatured
urease (red) on a bare HMDE at (B) different striping currents at
25 °C and (C) different temperatures using Istr −25 μA. Adapted with permission from
ref (238). Copyright
2013 Elsevier.
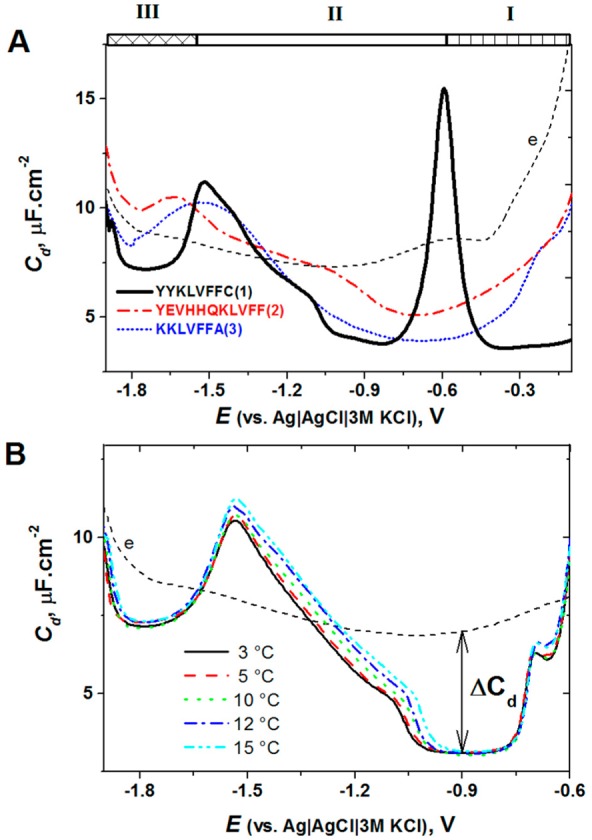
(A) Capacity–potential
curves of 1 μM peptide YYKLVFFC
(black), YEVHHQKLVFF (red), and KKLVFFA (blue) in 35 mM Na-phosphate,
pH 7 (dashed line). Peptides were adsorbed at −0.1 V, for tA of 120 s at HMDE, followed by recording of
ac voltammogram with a frequency of 150 Hz, amplitude of 5.0 mV, and
a scan rate of 8.0 mV/s. (B) Capacity–potential curves of 1
μM peptide YYKLVFFC recorded in 35 mM phosphate buffer, pH 7.0
at different temperatures, as indicated on the graph. Accumulation
potential EA of −0.5 V; accumulation
time tA of 120 s. Adapted with permission
from ref (221). Copyright
2013 Elsevier.
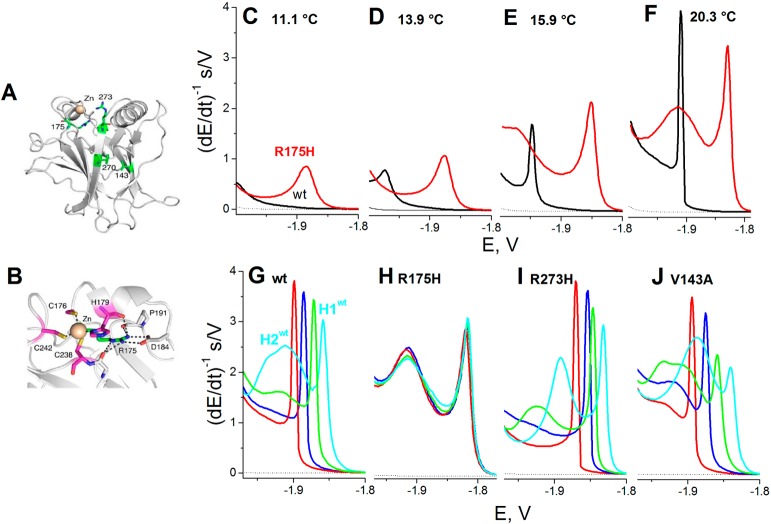
Structure of DNA-binding
domain of p53. (A) Overall structure of
T-p53C (PDB entry 1UOL). Sites of cancer mutations investigated
in this study (V143A, R175H, F270L, and R273H) are highlighted as
green stick models. (B) Close-up view of the zinc coordination sphere,
with the four zinc ligands shown in magenta. (C–F) CPS peak
H of wild type T-p53C (black) and mutant R175H (red) at DTT-HMDE in
50 mM phosphate, pH 7 at (C) 11.1 °C, (D) 13.9 °C, (E) 15.9
°C, and (F) 20.3 °C. (G–J) CPS peaks H of (G) wt,
(H) R175H, (I) R273H, and (J) V143A treated by 0 mM (red), 5 mM (blue),
10 mM (green), and 20 mM (cyan) EDTA at 0 °C for 10 min. CPS
measurements were performed at 18 °C. Adapted with permission
from ref (105). Copyright
2011 American Chemical Society.
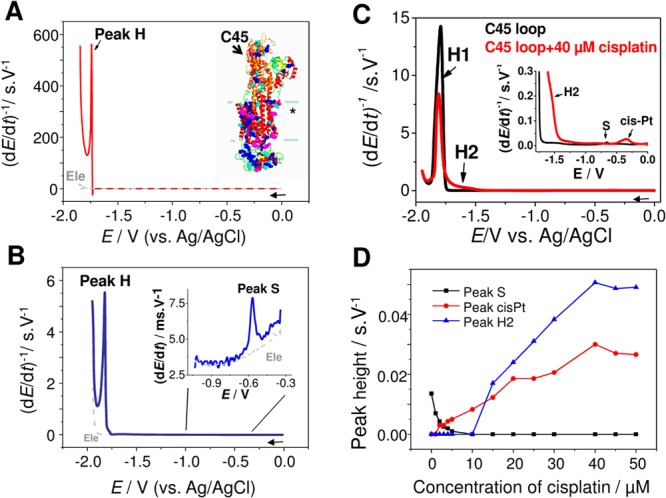
Chronopotentiograms of (A) Na+/K+-ATPase
(NKA) and (B) its C45 loop. CPS experiment at HMDE, concentration
of proteins: (A) 10 μM and (B) 500 nM, tA of 30 s; supporting electrolyte, Britton–Robinson
buffer, pH 6.5; stripping current Istr, −10 μA. (C) CPS records of 5 μM C45 loop before
(black) and after (red) incubation with cisplatin. (D) Dependence
of peak heights (C45 peak S, C45 peak H2, and cis-Pt) on the concentration
of cisplatin. Concentration of cisplatin (for C) was 40 μM,
activated complex was used for all experiments. EC parameters: supporting
electrolyte 0.2 M phosphate buffer, pH 7.4; tA of 30 s, Istr of −20 μA.
“*” in inset of panel A: NKA transmembrane part. Adapted
with permission from refs (410) and (412). Copyright 2012 Wiley-VCH Verlag GmbH&Co and 2012 Elsevier.
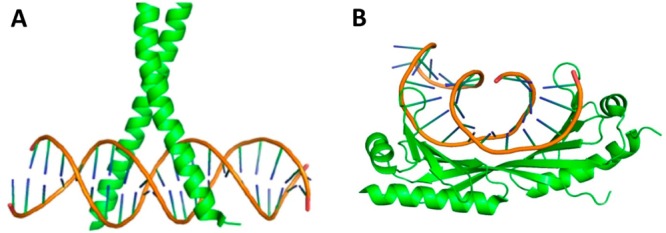
Different
DNA–protein binding modes. (A) Most proteins insert
helix element into the major groove of DNA molecule. For example,
helical bZIP motif of the AP-1 transcription factor binds within the
major groove of the specific DNA sequence (PDB: 1FOS). (B) Some proteins employ, however, β-sheets for their
binding into the minor groove of DNA. For example, TBP protein binds
to minor groove and partially unwinds and kinks DNA (PDB: 1YTB).
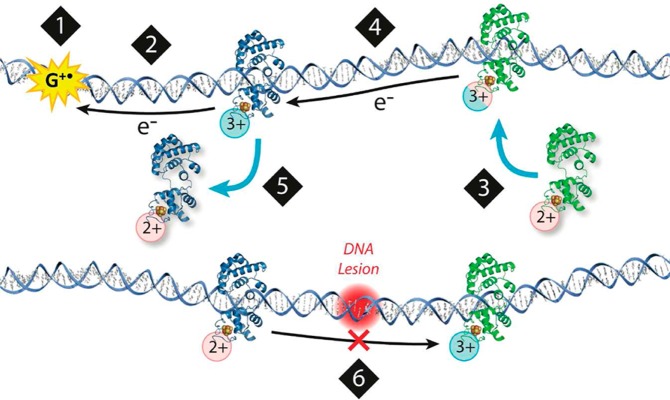
Model for a DNA-mediated search by repair proteins.
(1) When the
cell undergoes oxidative stress, guanine radicals are formed, triggering
a repair protein to bind DNA. (2) DNA-binding protein is oxidized,
releasing an electron that repairs the guanine radical. (3) Another
repair protein binds to a distant site. As it binds to DNA, there
is a shift in the redox potential of the protein, making it more easily
oxidized. (4) The protein could then send an electron through the
DNA base pair stack that travels to a distally bound protein, scanning
the intervening region for damage. (5) If the base pair stack is intact,
charge transport occurs between proteins. The repair protein that
receives the electron is reduced and dissociates. (6) If a lesion
is present (red), charge transport is attenuated, and the repair proteins
will remain bound in the oxidized form and slowly proceed to the site
of damage. Adapted with permission from ref (464). Copyright 2012 American
Chemical Society.
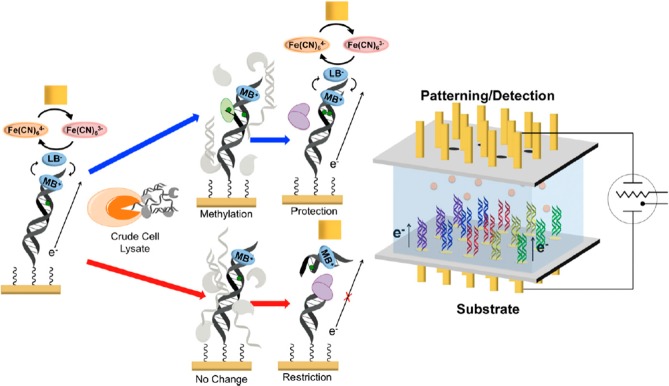
Electrochemical platform
(right) and scheme (left) for the detection
of human methyltransferase activity from crude cell lysates. (right)
The electrochemical detection platform contains two electrode arrays,
each with 15 electrodes (1 mm diameter each) in a 5 × 3 array.
Multiple DNAs are patterned covalently to the substrate electrode
by an electrochemically activated click reaction initiated with the
patterning electrode array. Once a DNA array is established on the
substrate electrode platform, electrocatalytic detection is then performed
from the top patterning/detection electrode. (left) Overview of electrochemical
detection scheme at each electrode of the 5 × 3 array. DNA, patterned
onto the bottom electrode using the copper-activated click chemistry,
is electrocatalytically detected from the top electrode using methylene
blue (MB+) as the electrocatalyst and ferricyanide for amplification.
Crude cell lysate is then added to the surface containing the patterned
DNA. If methyltransferase (green) is present (blue arrows), the hemimethylated
DNA on the electrode is methylated (green dot) by the methyltransferase
to a fully methylated duplex; if methyltransferase is not present
(red arrows), the hemimethylated DNA is not further methylated. A
methylation-specific restriction enzyme, BssHII (purple), is then
added. If the DNA is fully methylated (blue arrows), the electrochemical
signal remains protected, and the DNA is not cleaved. However, if
the DNA remains hemimethylated (red arrows), it is cut by the restriction
enzyme, and the electrocatalytic signal associated with MB+ binding
to DNA is diminished significantly. Adapted with permission from ref (479). Copyright 2014 Proceedings
of the National Academy of Sciences of the United States of America.
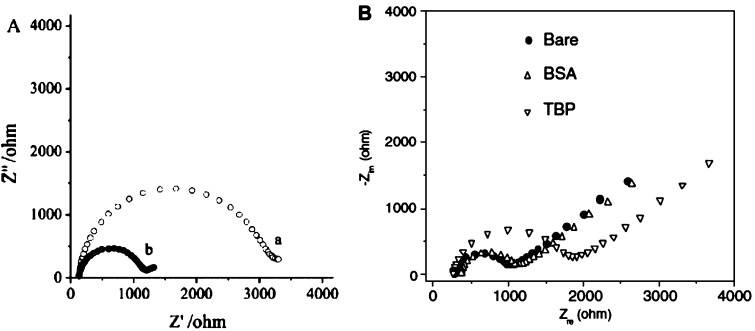
(A) Impedance spectra
of a gold electrode with NF-κB-specific
dsDNA before (a) and after (b) incubation with 33 μg/mL NF-κB
p50, as measured by Tersch and Lisdat. (B) Nyquist plots of gold electrode modified with DNA duplexes before
and after interaction with BSA and TBP, as reported by Chang and Li. Electrolyte: 10 mM PBS (pH 7.0) with 10 mM
[Fe(CN)6]3–/4– and 10 mM NaCl.
Frequency: from 0.1 Hz to 100 k Hz. Amplitude: 10 mV. Bias potential:
0.20 V. Adapted with permission from refs (485) and (484). Copyright 2011 and 2009 Elsevier.
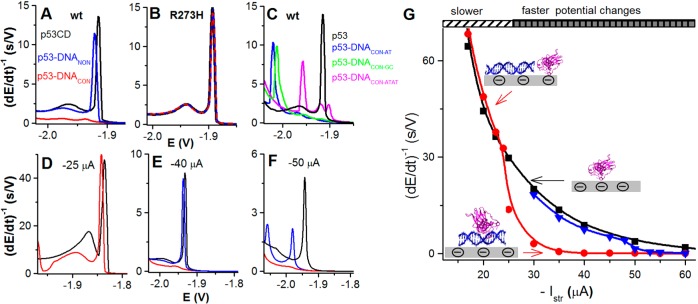
(A) Sequence-specific
binding of wild-type p53 core domain (p53CD)
to dsDNACON as detected by CPS at a DTT-modified hanging
mercury drop electrode (DTT-HMDE) using Istr −35 μA at 21 °C. Free p53CD (black), sequence-specific
p53CD–DNACON complex (red), and a mixture of p53CD
with 40-mer dsDNA not containing the consensus sequence (blue, p53CD
+ dsDNANON). (B) Interaction of mutant p53CD R273H with
dsDNACON (showing no DNA binding). Free p53CD R273H (black),
p53CD R273H + dsDNACON (red), p53CD R273H + dsDNANON (blue). (C) Peak H of p53CD (black) and p53CD complexes with spacer-containing
DNAs: DNACON–GC (green), DNACON–AT (blue), and DNACON–ATAT (magenta); Istr −35 μA at 21 °C. (D–F) Peak
H of p53CD (black), p53CD–DNACON complex (red),
and p53CD–DNANON (blue) at Istr of (D) −25 μA, (E) −40 μA, and
(F) −50 μA. (G) Dependence of peak H1 height of free
p53CD (black), p53CD–DNACON complex (red), and p53CD–DNANON (blue) on stripping current (−Istr). Adapted with permission from ref (240). Copyright 2014 Elsevier.
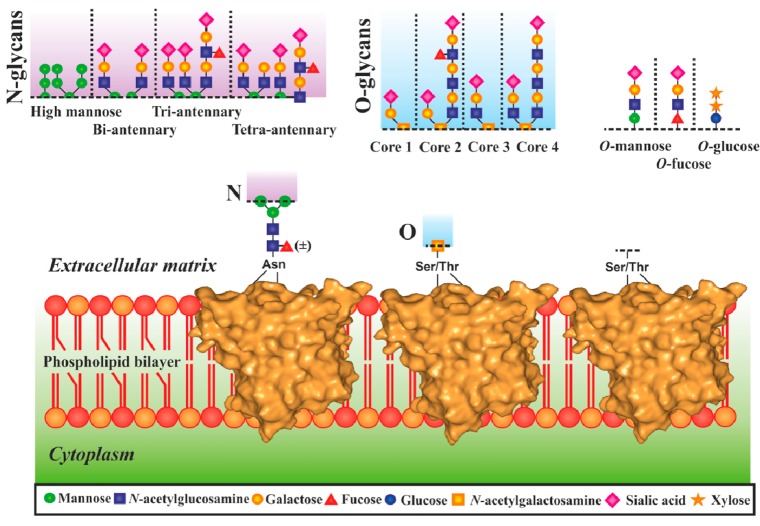
Graphical
representation of complexity of glycans, showing variability
of sugar building blocks, multiple branching, and attachment points.
Adapted with permission from ref (37). Copyright 2013 Springer Science and Business
Media.
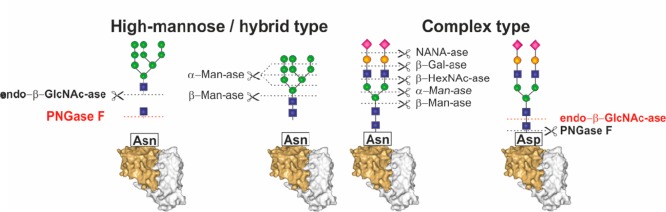
Enzymatic release of whole glycans or carbohydrates
from glycoproteins.
Abbreviation of enzymes: endo-β-GlcNAc-ase, endo-β-N-acetylglucosaminidase (endo H); PNGase F, peptide:N-glycosidase F (PNGase F); α-Man-ase, α-mannosidase;
β-Man-ase, β-mannosidase; NANA-ase, N-acetylneuraminic acid hydrolase (sialidase); β-Gal-ase, β-galactosidase;
and β-HexNac-ase, β-N-acetylhexosaaminidase.
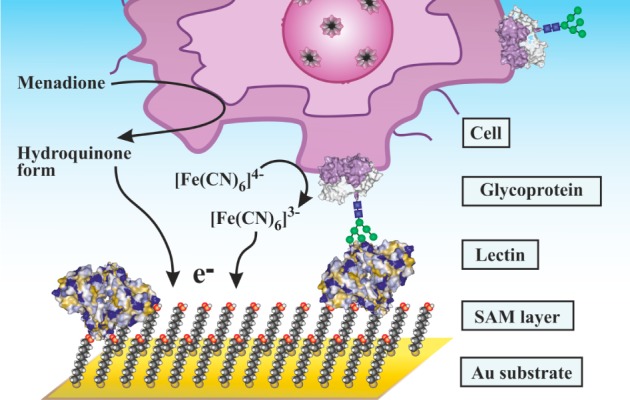
Schematic representation of EC detection of
cells with lectins
by monitoring of respiratory activity of the cells with a two-mediator
system.
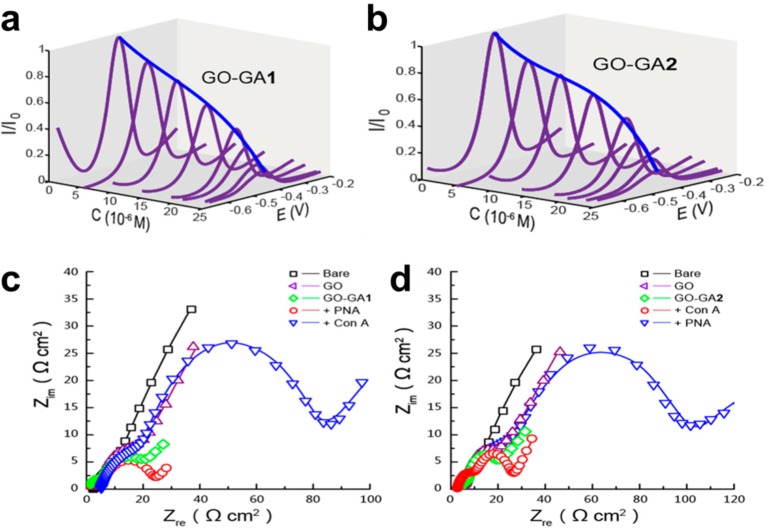
Probing the specific sugar–lectin interactions on surface
modified by (a,c) glucose and (b,d) galactose. The interaction between
lectin and immobilized saccharide was detected by either (a,b) DPV
or (c,d) EIS. Bare screen printed electrode was modified by graphene
oxide (GO), then by antraquinone containing glucose (GO-GA1) or galactose
(GO-GA2), and interaction between saccharide-containing surfaces was
probed by two lectins: Con A, recognizing glucose; and peanut agglutinin,
recognizing galactose. All experiments were performed in Tris-HCl
(pH 7.0). Adapted with permission from ref (634). Copyright 2013 Macmillan Publishers Ltd.:
Scientific Reports.
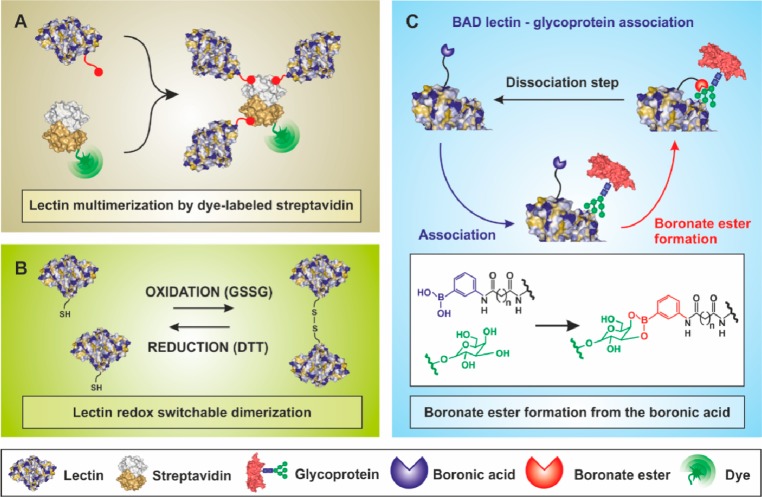
Various ways to prepare modified lectins with improved
binding
properties. (A) Modification of a lectin by a biotin derivative, which
upon incubation with a dye-labeled streptavidin forms a lectin multimer.
Adapted with permission from ref (637). Copyright 2013 American Chemical Society.
(B) A redox switchable formation of a lectin dimer involving a thiolated
form of a lectin. Adapted with permission from ref (638). Copyright 2004 John
Wiley & Sons. (C) A scheme of increased strength of interaction
between BAD (boronic acid-decorated) lectin and a glycan with involvement
of lectin binding site and boronate derivative in the biorecognition.
Adapted with permission from ref (639). Copyright 2013 American Chemical Society.
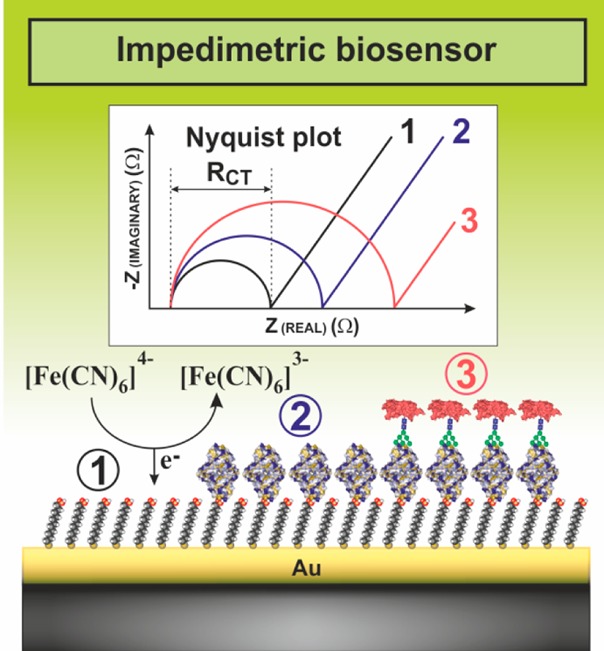
A typical
interfacial layer of the EIS-based biosensor after SAM
formation (1), immobilization of lectins (2), and biorecognition of
a glycoprotein (3) with corresponding Nyquist plots showing shifts
in the RCT value with an increased loading
of the surface.

AFM images
of the gold surfaces during a patterning procedure starting
with the bare gold (upper left), the gold surface modified by a mixed
SAM (upper right), the surface with covalently attached SNA I lectin
(lower left), and the surface after being treated with a blocking
agent (lower right). Scale of z-axis was adjusted
in a way to clearly see topological features on the surface after
each modification step. Adapted with permission from ref (653). Copyright 2013 Springer
Science and Business Media.
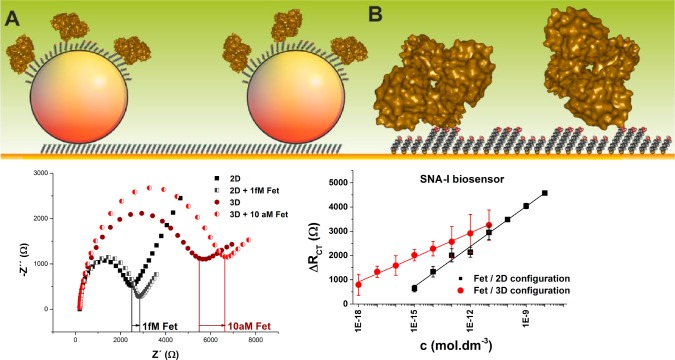
A graphical
representation drawn to scale of interfaces applied
to build (A) the 3D biosensor based on integrated 20 nm gold nanoparticles
or (B) the 2D biosensor (upper image). The 3D biosensor was built
on a planar gold surface by chemisorptions of 11-aminoundecanethiol
for attachment of 20 nm gold nanoparticles (spheres). (A) On every
gold nanoparticle, a mixed SAM composed of 11-mercaptoundecanoic acid
(MUA) and 6-mercaptohexanol was formed for covalent immobilization
of lectin. (B) The 2D biosensor was formed by incubation of a planar
gold with MUA and mercaptohexanol for covalent attachment of a lectin.
In the lower part of the figure, comparison of the response of the
2D and the 3D biosensor to its analyte fetuin (Fet) with concentration
close to the LOD, represented in a Nyquist plot (left), and calibration
graphs for detection of fetuin (Fet) by both biosensors (right) are
shown. Adapted with permission from ref (655). Copyright 2014 Enterprise Strategy Group.
(A,B) CPS and (C) SWV
curves of chitosan at mercury electrodes.
(A,C) 10 μg/mL of chitosan at HMDE and (B) 15 μg/mL of
chitosan at solid amalgam electrode. Accumulation time, tA, 60 s; stripping current intensity, (A) Istr, −70 μA; (B) Istr, −40 μA; (C) frequency 20 Hz; (D) CPS curves of 12
μM chitohexaose (red) and N,N′,N″,N‴,N‴′,N‴″-hexaacetylchitohexaose
(blue); tA, 60 s; Istr, −40 μA. Background: 0.1 M sodium acetate,
pH 5.2 (dashed). Adapted with permission from ref (246). Copyright 2014 Elsevier.
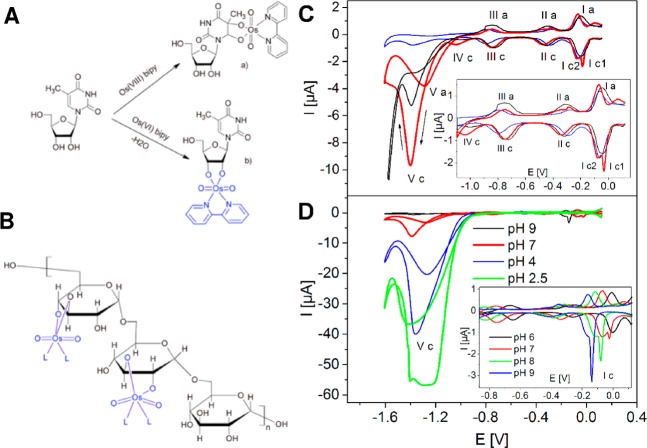
(A) Reaction of Os(VIII)L and Os(VI)L complexes with different
parts of a nucleoside showing Os(VI)L complex specifically modifying
ribose moiety. (B) Fragment of Os(VI)L-modified dextran. (C) Adsorptive
stripping cyclic voltammograms of 10 mM base-modified (blue) and sugar-modified
thymine riboside (red), and sugar-modified adenine riboside (black).
(D) Dependence on pH, 10 mM sugar-modified thymine riboside, HMDE,
with stirring; Britton–Robinson buffer, pH 7.0; scan rate (C)
2 V/s, (D) 1 V/s; tA of 60 s; EA of 0 V, step potential 5 mV. Adapted with
permission from ref (694). Copyright 2007 John Wiley & Sons, Inc.
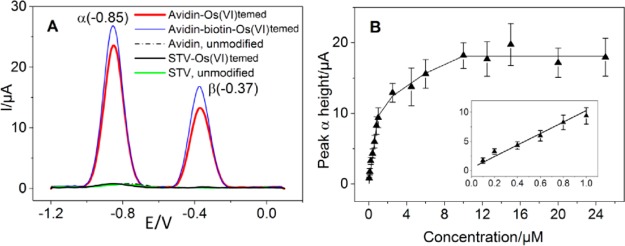
(A) AdTS SWV of unpurified 5 μM Os(VI)temed-treated
avidin,
complex avidin–biotin, and streptavidin (STV); (B) concentration
dependence of peak α (EP ≈
−0.85 V) of avidin-Os(VI)temed, four replicative measurements.
Inset: Detail of the concentration range 100–1000 nM. Adapted
with permission from ref (702). Copyright 2014 Elsevier.
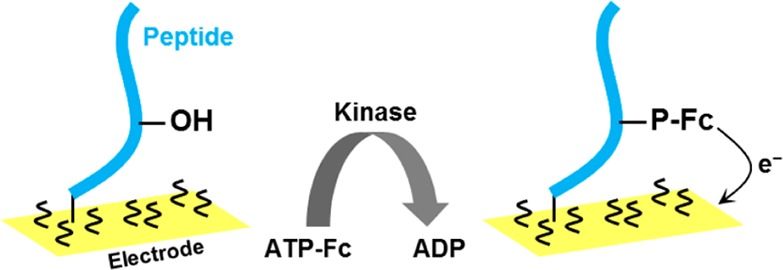
Protein kinase C-catalyzed phosphorylation
of SIYRRGS RRWRKL peptide (with phosphorylated
serine underlined)
using ferrocene-labeled ATP (ATP-Fc) as a substrate. After a transfer
of γ-phosphate-Fc group to the serine residue of the peptide,
the surface-attached Fc groups are detected via EC techniques at thiol-modified
gold electrodes. Adapted with permission from ref (754). Copyright 2008 Royal
Society of Chemistry.
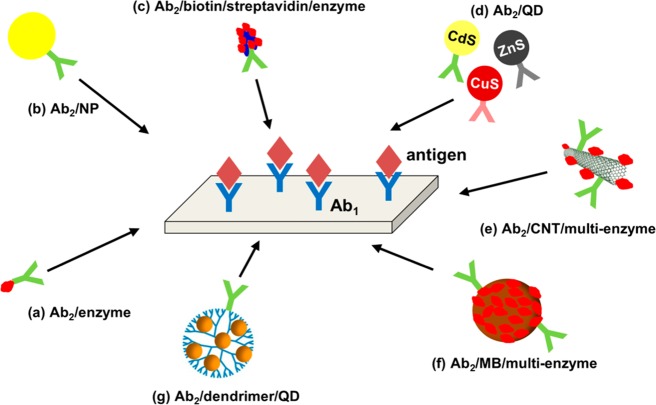
Scheme of amplification
strategies for detection of protein cancer
biomarkers in EC immunoassays. Surface-immobilized primary antibody
(Ab1) captures an antigen, which is then detected using,
for example, (a) a simple secondary antibody/enzyme (Ab2/enzyme) or (b) antibody/nanoparticle (Ab2/NP) bioconjugate,
which are now often replaced with more sophisticated systems, in which
the secondary antibody is coupled with, for example, (c) biotin/streptavidin/enzyme, (d) different quantum dots (Ab2/QD), (e) enzyme-modified carbon nanotubes (Ab2/CNT/multienzyme), (f) magnetic
beads bearing cluster of enzymes (Ab2/MB/multienzyme), or (g) quantum dot-dendrimer nanocomposites
(Ab2/dendrimer/QD).
Examples
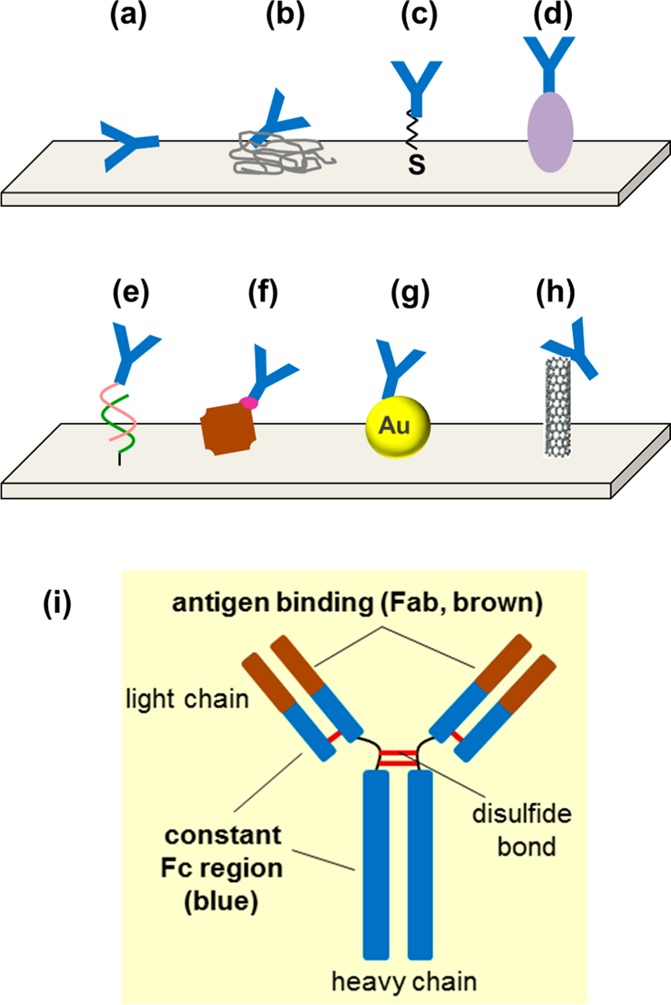
of antibody immobilization to the surface. The binding
can be achieved via, for example, (a) physical adsorption, (b) entrapment
into a polymer matrix, (c) thiol groups, (d) protein A or G, (e) DNA-directed
immobilization (by site-specific coupling of protein G to DNA oligonucleotide),
(f) avidin–biotin system, (g) nanoparticle, or (h) carbon nanotubes
(linked via carboxyl groups at CNT). (i) Structure of the antibody
with Fab and Fc region.
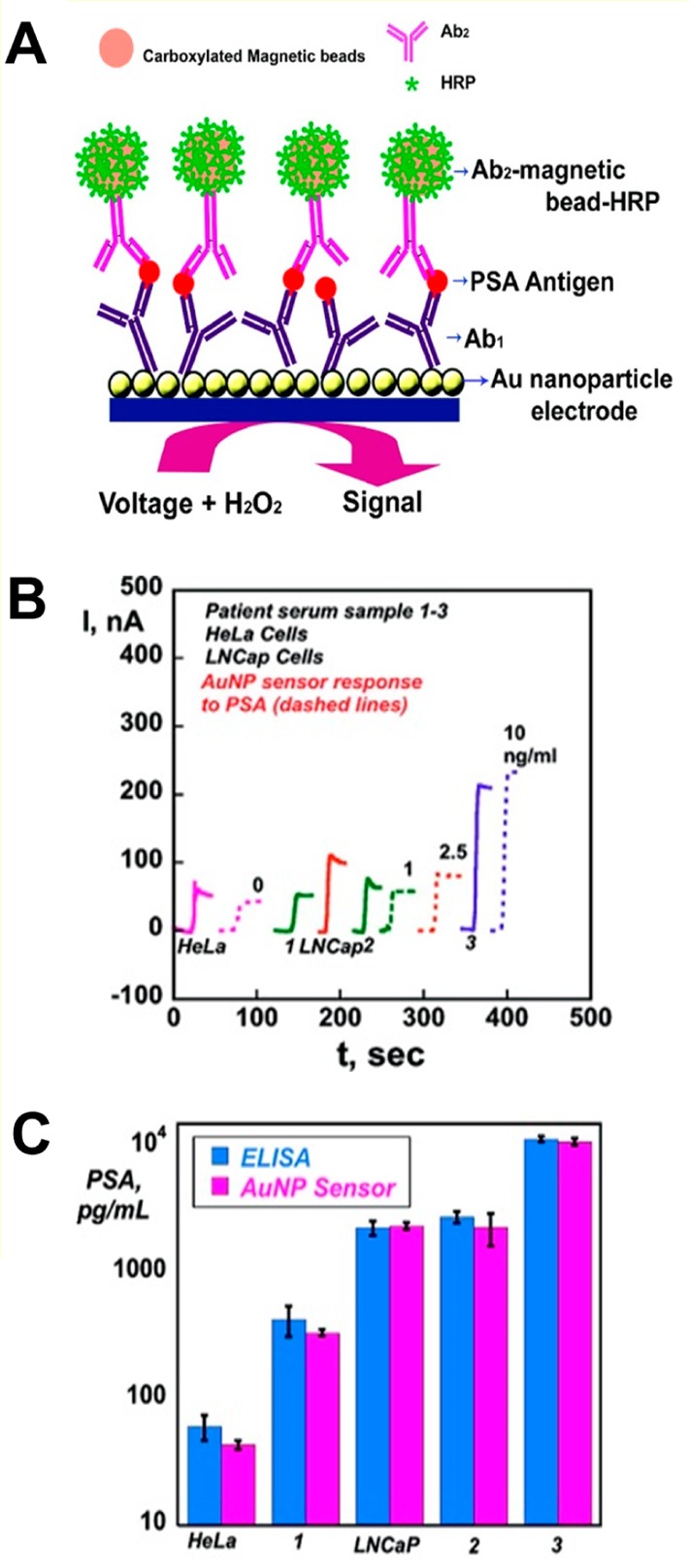
(A) Gold nanoparticle
(AuNP)-based immunosensor. The immunosensor
involves attached Ab1, which captures antigen from a sample,
followed by incubation with Ab2-magnetic bead-HRP (Ab2-MB-HRP), providing multiple enzyme labels for each PSA bound.
The detection step involves immersing the immunosensor into a buffer
containing a mediator, applying voltage, and injecting H2O2. (B) Results for AuNP immunosensor incubated with PSA
present in 10 μL of a calf serum (ng/mL labeled on curves, dashed
lines), cell lysates (HeLa and LNCap cells), and human patient serum
samples (1–3) (solid lines) for 1.25 h, followed by an injection
of 10 μL of 4 pmol/mL of Ab2-MB-HRP. (C) Validation
of AuNP sensor results for cell lysate and human serum samples by
comparing against results from an ELISA determination (relative standard
deviation ∼10%) for the same samples. Adapted with permission
from ref (794). Copyright
2009 American Chemical Society.
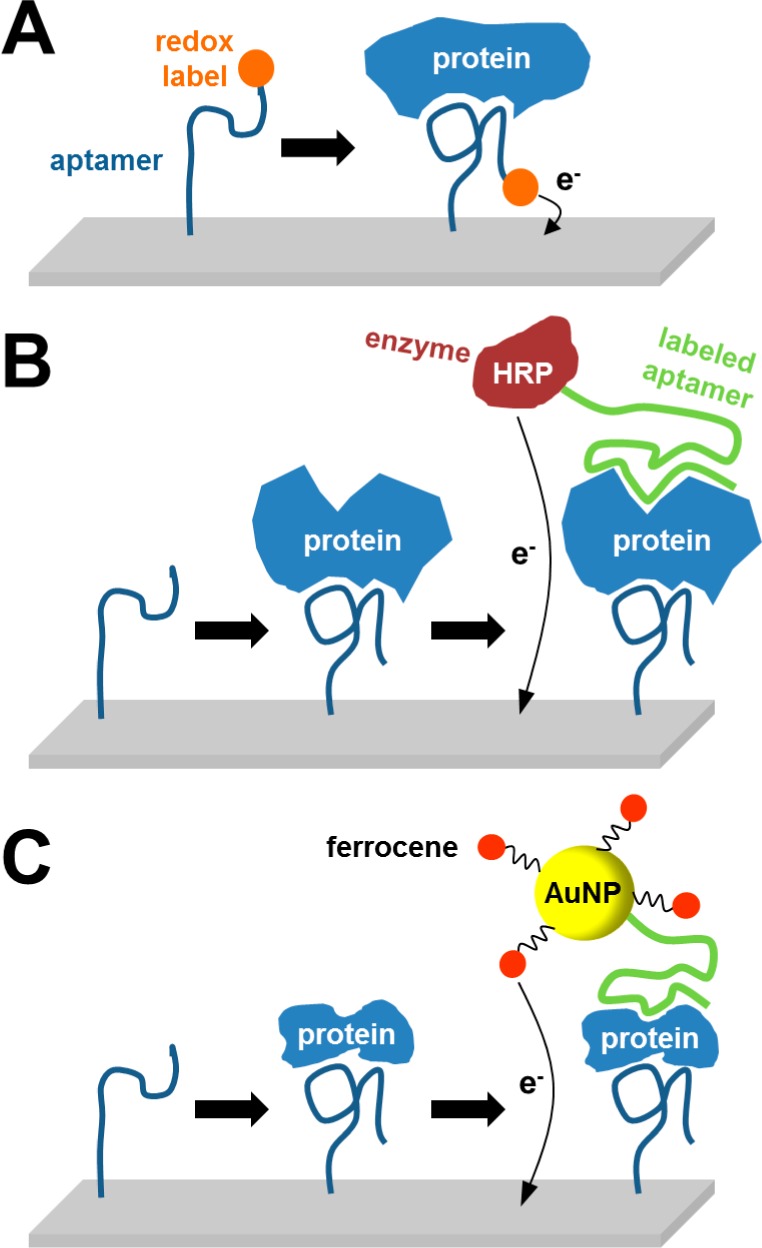
Different approaches for detection of proteins
using nucleic acid
aptamers. (A) Redox-labeled aptamer alters its conformation after
aptamer–protein complex formation, positioning the label closer
to the electrode. (B) Strategy employing
two aptamers, electrode-immobilized aptamer for capturing the protein
and a second aptamer labeled with enzyme for EC monitoring of enzymatic
reaction. (C) Approach similar to that
in (B), with the second aptamer being labeled with gold nanoparticle–ferrocene
conjugate.
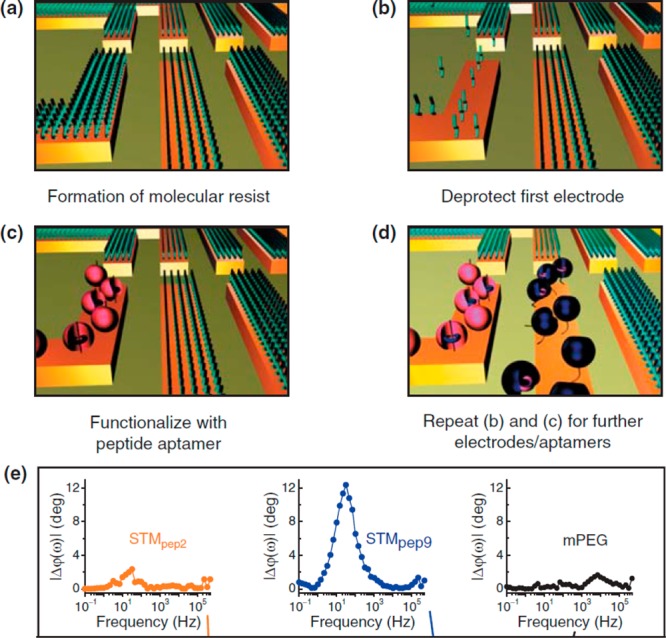
Electrochemically triggered
immobilization of peptide aptamers
within a biochip. (a) All microelectrodes are initially protected
by a mask from mPEG, resisting protein adsorption. (b) Release of
a mask from the electrode #1 by a highly negative voltage. (c) Funcionalization
of the electrode #1 with a peptide aptamer. (d) Independent functionalization
of the whole array by various peptide aptamers by repeating steps
(a)–(c). (e) Analysis of CDK2 bound to pep2, pep9, or to a
reference surface covered by mPEG as change in a phase shift, ϕ(ω),
which is a phase difference between an applied working potential and
measured current. A typical dependence of ϕ(ω) as a function
of applied frequency ω used in the analysis is shown. mPEG is
a methyl-terminated polyethylene glycol containing thiol. Reprinted
with permission from ref (832). Copyright 2008 BioMed Central.
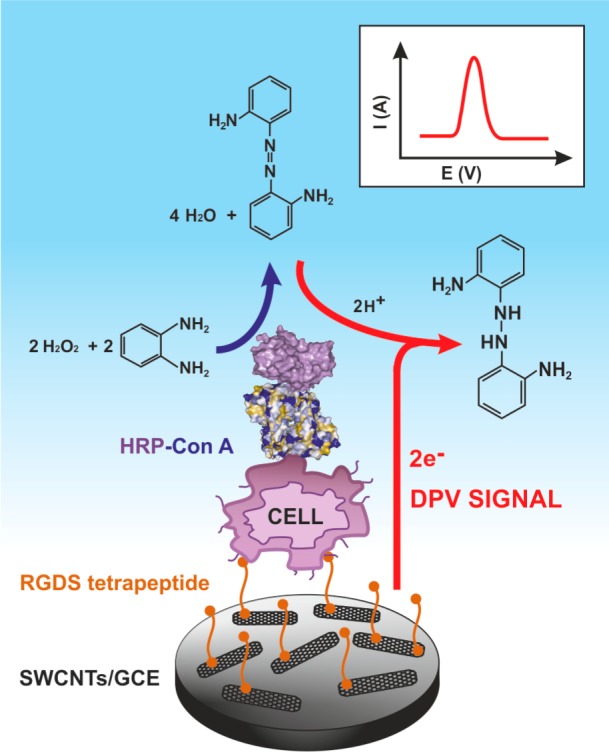
Analysis
of a gastric carcinoma cell line after a nonspecific attachment
of cells to a RGDS peptide. In the following step, a Con A–HRP
conjugate was injected to probe glycans on the cell surface. A DPV
signal was acquired by analysis of a product of oxidation of o-phenylendiamine by HRP in the presence of hydrogen peroxide.
Adapted with permission from ref (860). Copyright 2008 American Chemical Society.
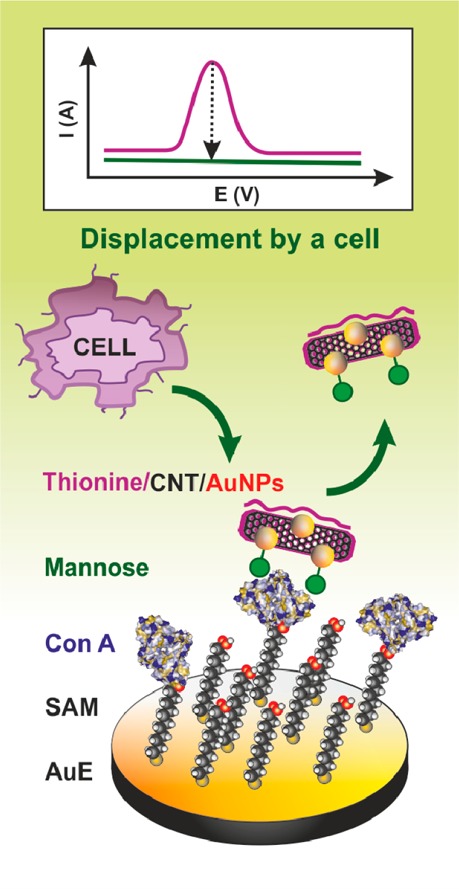
A displacement strategy for analysis of cells. Con A was
immobilized
on SAM layer by covalent coupling. A thionine/carbon nanotube (CNT)/gold
nanoparticles (AuNPs) nanocomposite was then incubated with the biosensor,
and displacement of the nanocomposite from the surface of the Con
A biosensor after incubation with cells resulted in a decrease of
EC response. Adapted with permission from ref (870). Copyright 2011 Royal
Society of Chemistry.
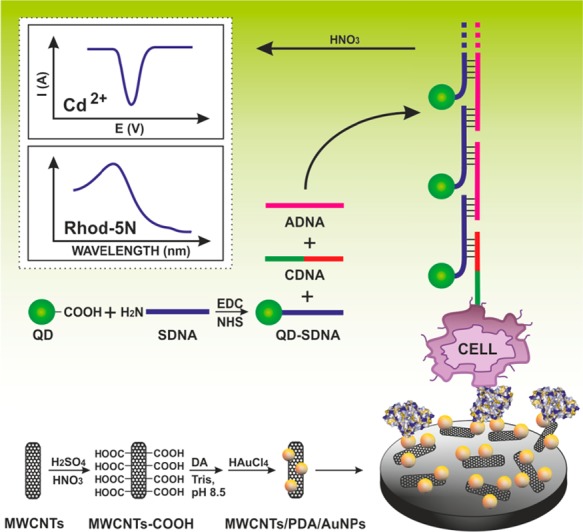
A supersandwich strategy for signal amplification in cancer cell
analysis. Initially, GCE was modified by oxidized MWCNTs, dopamine,
and AuNPs to which Con A was immobilized for detection of a CCRF-CEM
cancerous cell line. DNA concatamer was first assembled from a capture
DNA-aptamer (CDNA), a CdTe QD-labeled signal DNA (SDNA), and an auxiliary
DNA (ADNA). When the cells were attached to the surface, they were
probed by a preassembled concatamer, and finally a binding event was
detected by an anodic stripping voltammetry of Cd2+. Adapted
with permission from ref (873). Copyright 2013 American Chemical Society.
Similar articles
-
Recent Progress in Electrochemical Biosensors for Glycoproteins.Sensors (Basel). 2016 Dec 1;16(12):2045. doi: 10.3390/s16122045. Sensors (Basel). 2016. PMID: 27916961 Free PMC article. Review.
-
Electrochemical sensors and biosensors.Anal Chem. 2012 Jan 17;84(2):685-707. doi: 10.1021/ac202878q. Epub 2011 Nov 11. Anal Chem. 2012. PMID: 22044045 Free PMC article. Review. No abstract available.
-
Electrochemical biosensors on platforms of graphene.Chem Commun (Camb). 2013 Oct 25;49(83):9526-39. doi: 10.1039/c3cc44735a. Chem Commun (Camb). 2013. PMID: 24025792 Review.
-
Electrochemical sensors and biosensors based on nanomaterials and nanostructures.Anal Chem. 2015 Jan 6;87(1):230-49. doi: 10.1021/ac5039863. Epub 2014 Dec 19. Anal Chem. 2015. PMID: 25354297 Free PMC article. Review. No abstract available.
-
New electrochemical methods.Anal Chem. 2012 Jan 17;84(2):669-84. doi: 10.1021/ac2026767. Epub 2011 Nov 9. Anal Chem. 2012. PMID: 22017638 Review. No abstract available.
Cited by
-
Full-length antibodies versus single-chain antibody fragments for a selective impedimetric lectin-based glycoprofiling of prostate specific antigen.Electrochim Acta. 2017 Aug 20;246:399-405. doi: 10.1016/j.electacta.2017.06.065. Electrochim Acta. 2017. PMID: 29104305 Free PMC article.
-
Biosensors in Health Care: The Milestones Achieved in Their Development towards Lab-on-Chip-Analysis.Biochem Res Int. 2016;2016:3130469. doi: 10.1155/2016/3130469. Epub 2016 Mar 3. Biochem Res Int. 2016. PMID: 27042353 Free PMC article. Review.
-
Electrochemical detection of different p53 conformations by using nanostructured surfaces.Sci Rep. 2019 Nov 22;9(1):17347. doi: 10.1038/s41598-019-53994-6. Sci Rep. 2019. PMID: 31758050 Free PMC article.
-
Aberrant sialylation of a prostate-specific antigen: Electrochemical label-free glycoprofiling in prostate cancer serum samples.Anal Chim Acta. 2016 Aug 31;934:72-9. doi: 10.1016/j.aca.2016.06.043. Epub 2016 Jul 1. Anal Chim Acta. 2016. PMID: 27506346 Free PMC article.
-
Graphene as a signal amplifier for preparation of ultrasensitive electrochemical biosensors.Chem Zvesti. 2015 Jan;69(1):112-133. doi: 10.1515/chempap-2015-0051. Epub 2014 Nov 28. Chem Zvesti. 2015. PMID: 27242391 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
