

N17 Modifies mutant Huntingtin nuclear pathogenesis and severity of disease in HD BAC transgenic mice
- PMID: 25661181
- PMCID: PMC4386927
- DOI: 10.1016/j.neuron.2015.01.008
N17 Modifies mutant Huntingtin nuclear pathogenesis and severity of disease in HD BAC transgenic mice
Abstract
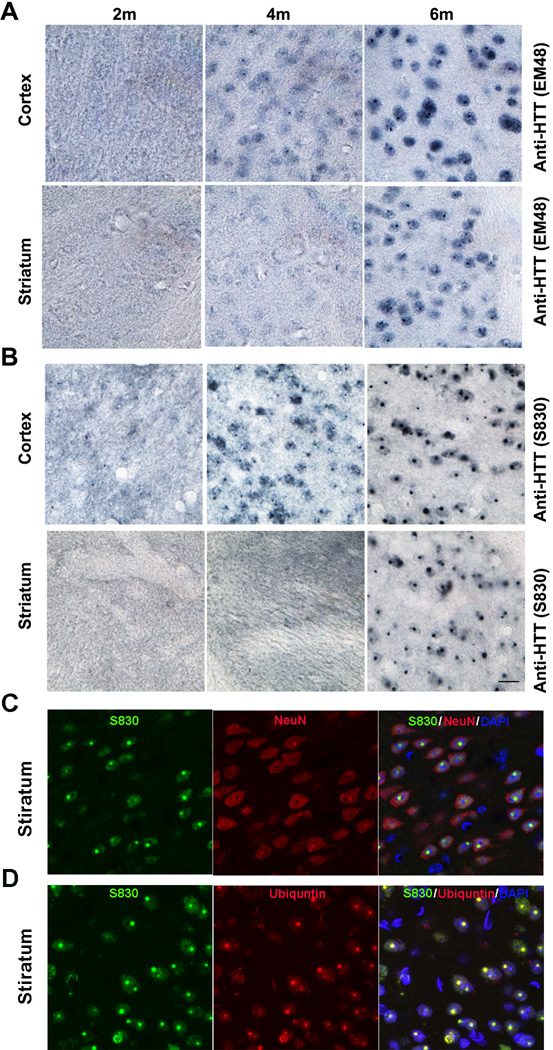
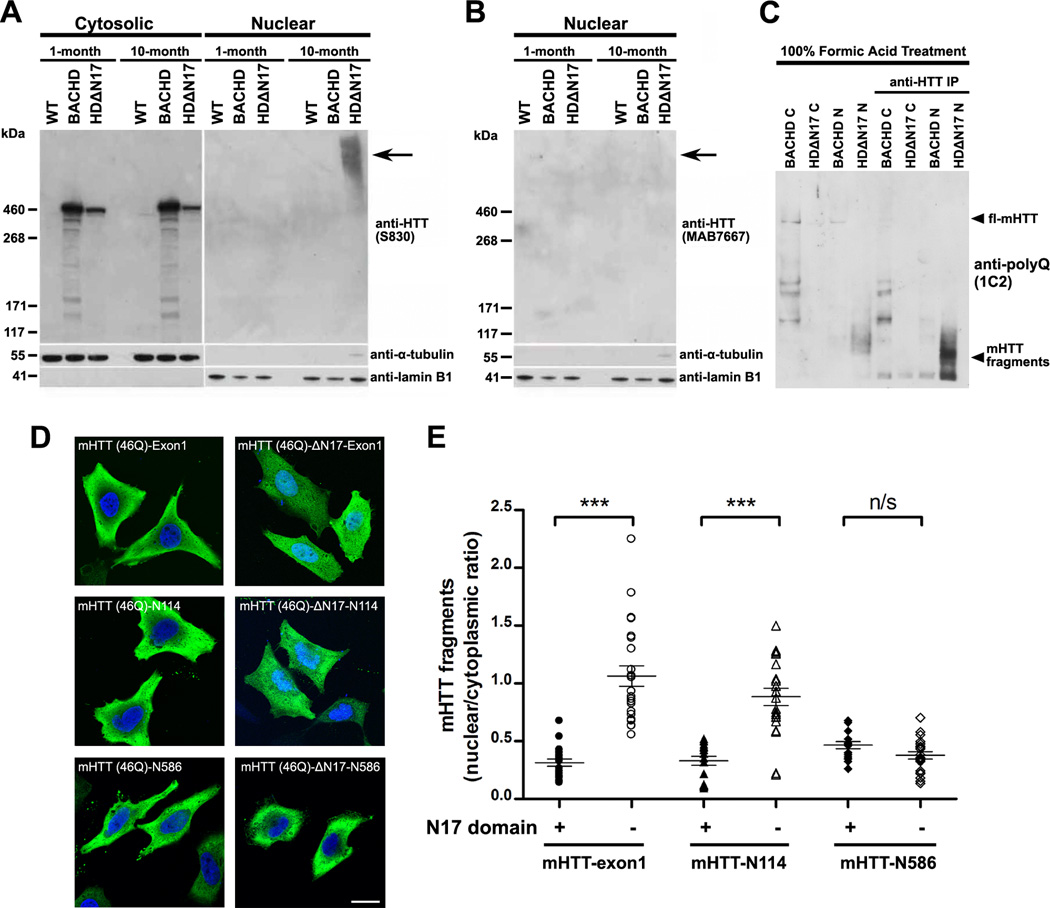
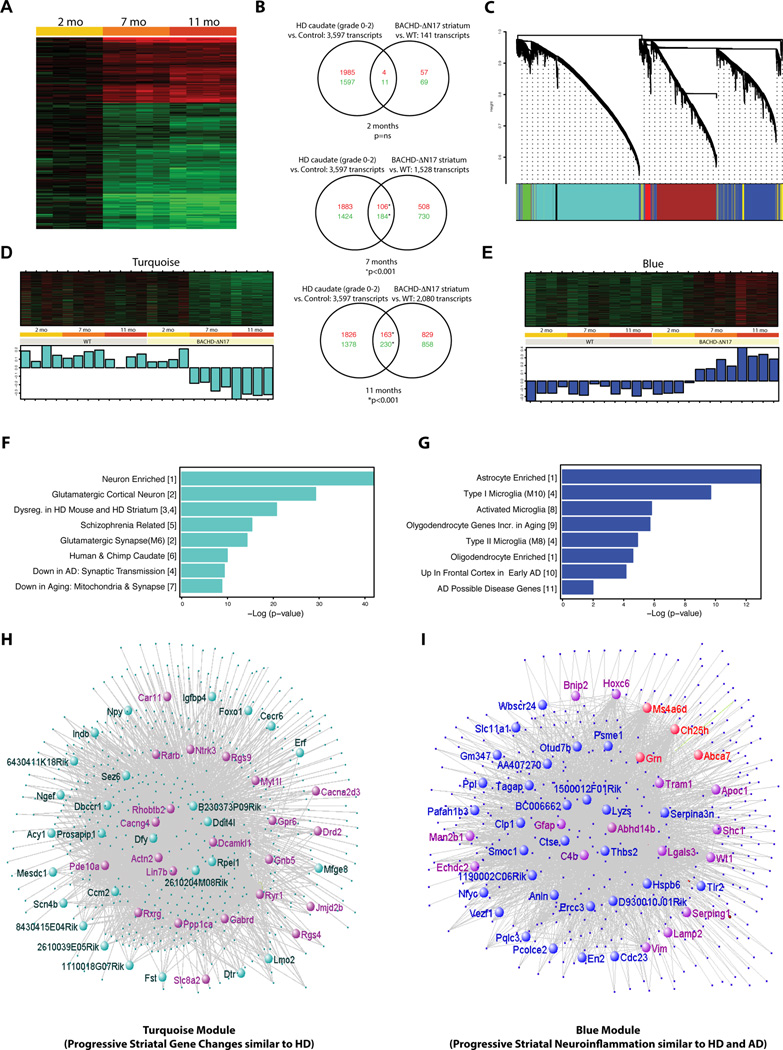
The nucleus is a critical subcellular compartment for the pathogenesis of polyglutamine disorders, including Huntington's disease (HD). Recent studies suggest the first 17-amino-acid domain (N17) of mutant huntingtin (mHTT) mediates its nuclear exclusion in cultured cells. Here, we test whether N17 could be a molecular determinant of nuclear mHTT pathogenesis in vivo. BAC transgenic mice expressing mHTT lacking the N17 domain (BACHD-ΔN17) show dramatically accelerated mHTT pathology exclusively in the nucleus, which is associated with HD-like transcriptionopathy. Interestingly, BACHD-ΔN17 mice manifest more overt disease-like phenotypes than the original BACHD mice, including body weight loss, movement deficits, robust striatal neuron loss, and neuroinflammation. Mechanistically, N17 is necessary for nuclear exclusion of small mHTT fragments that are part of nuclear pathology in HD. Together, our study suggests that N17 modifies nuclear pathogenesis and disease severity in HD mice by regulating subcellular localization of known nuclear pathogenic mHTT species.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
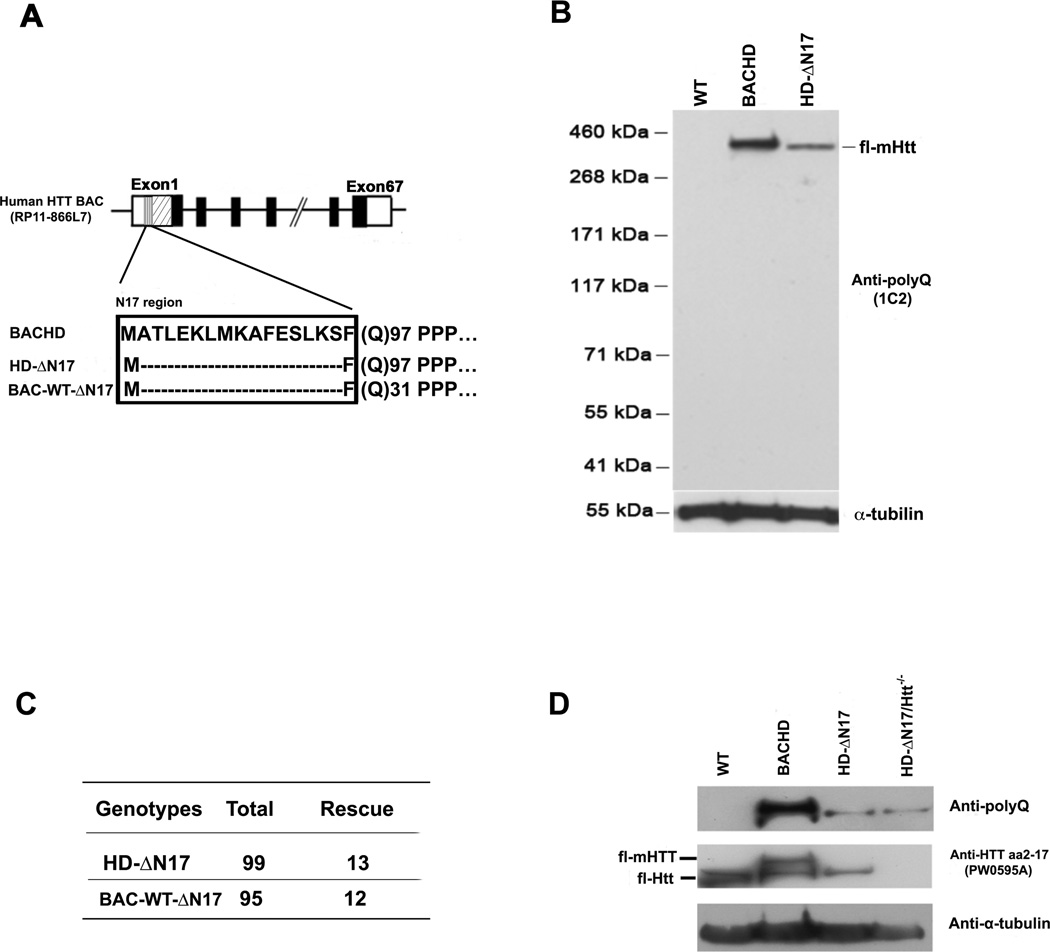
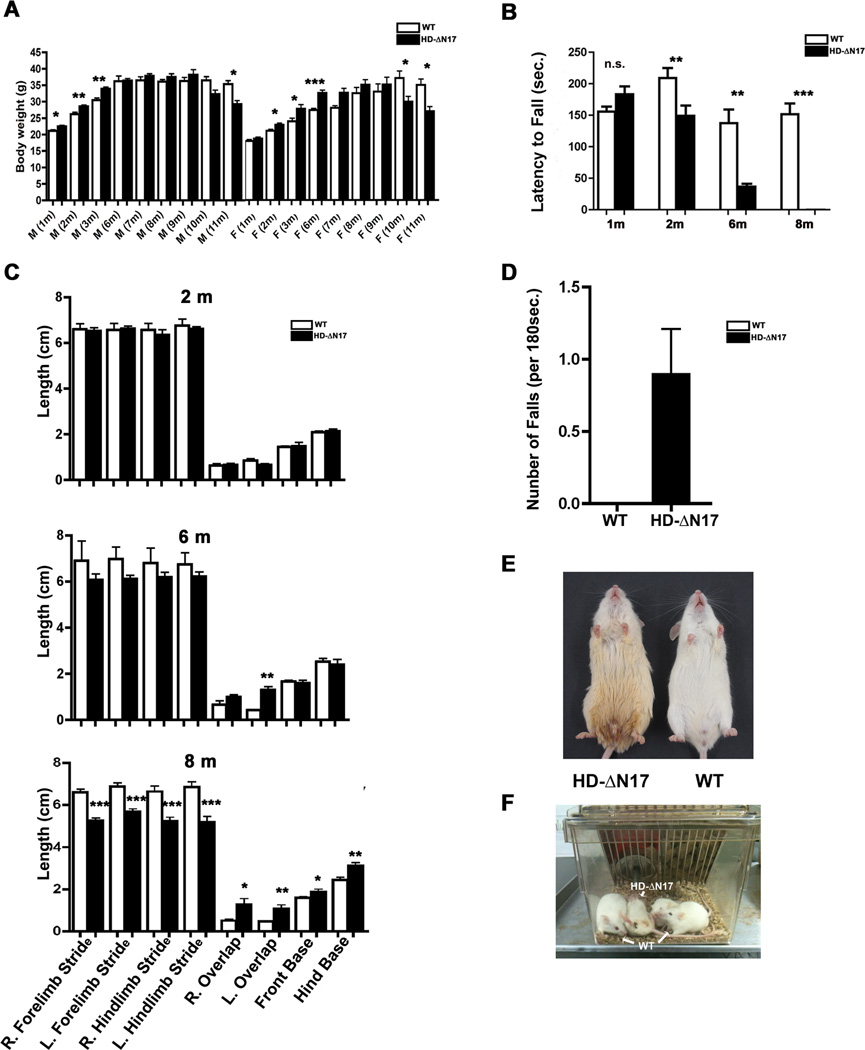
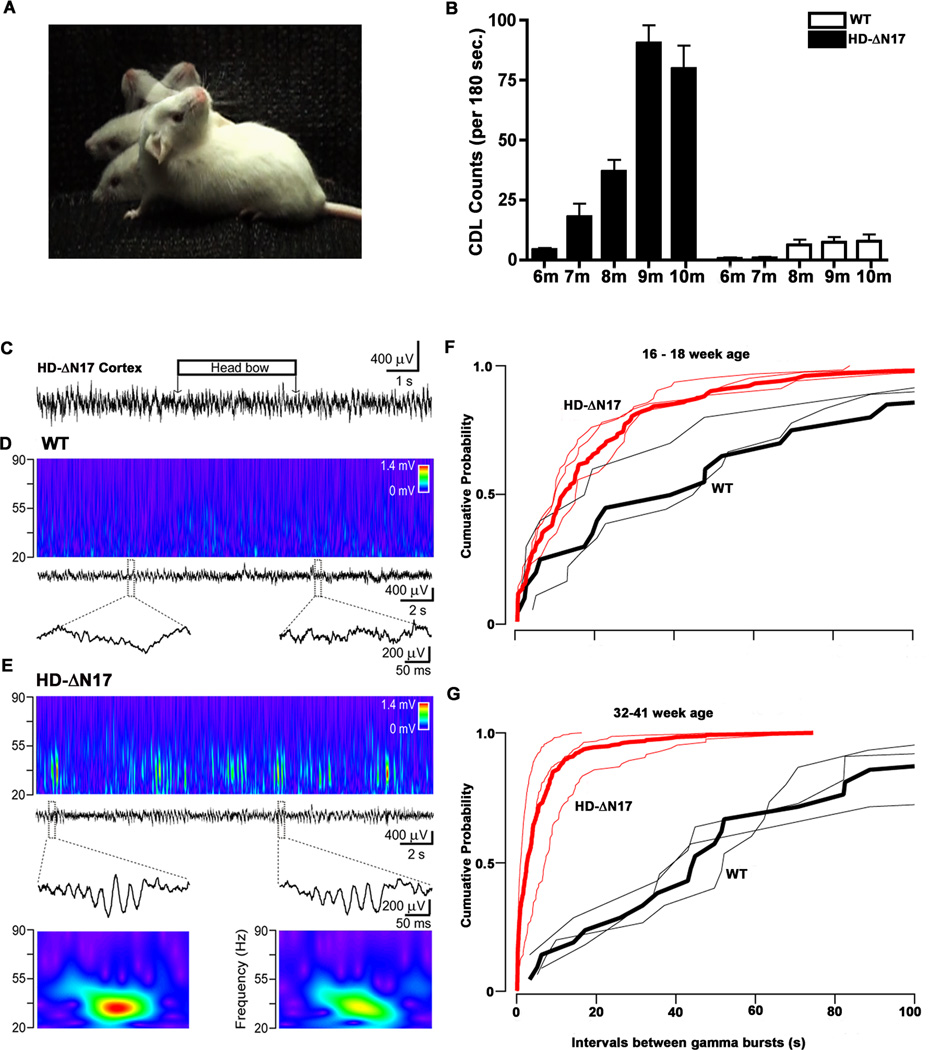
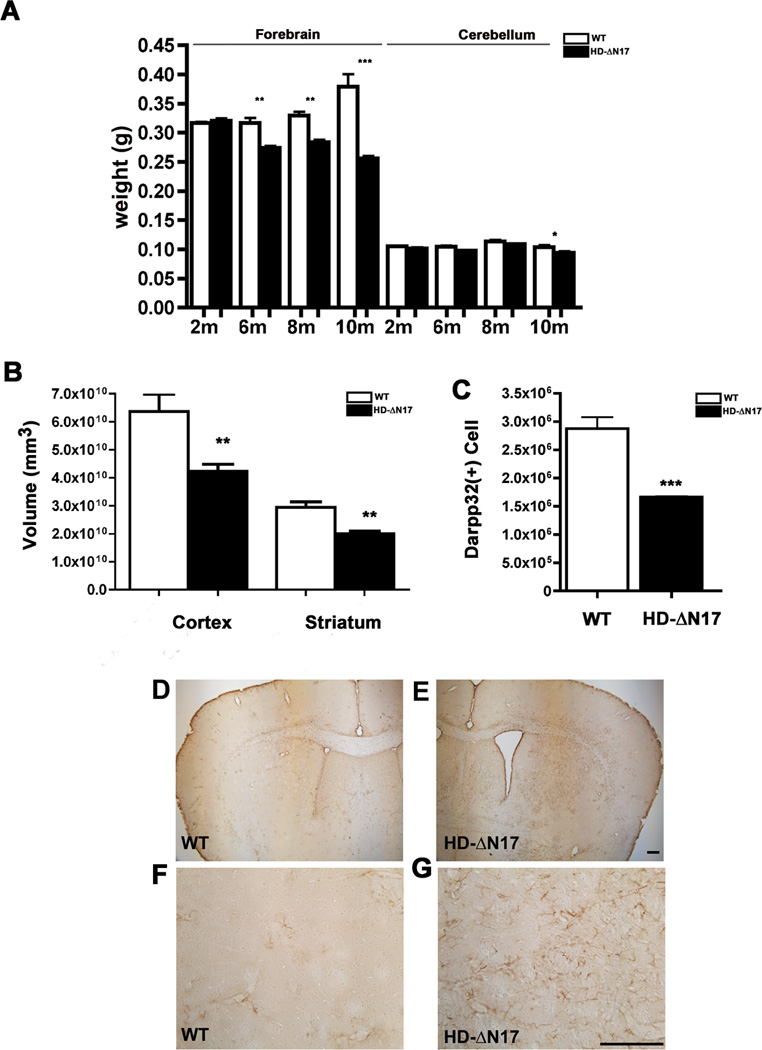

References
-
- Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, Truant R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nature Chemical Biology. 2011;7:453–452. - PubMed
-
- Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Human Molecular Genetics. 2007;16:2600–2615. - PubMed
-
- Benn CL, Landles C, Li H, Strand AD, Woodman B, Sathasivam K, Li SH, Ghazi-Noori S, Hockly E, Faruque SM, et al. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington's disease. Hum Mol Genet. 2005;14:3065–3078. - PubMed
-
- Burgess DL, Noebels JL. Single gene defects in mice: the role of voltage-dependent calcium channels in absence models. Epilepsy Res. 1999;36:111–122. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
