

A global profile of replicative polymerase usage
- PMID: 25664722
- PMCID: PMC4789492
- DOI: 10.1038/nsmb.2962
A global profile of replicative polymerase usage
Abstract
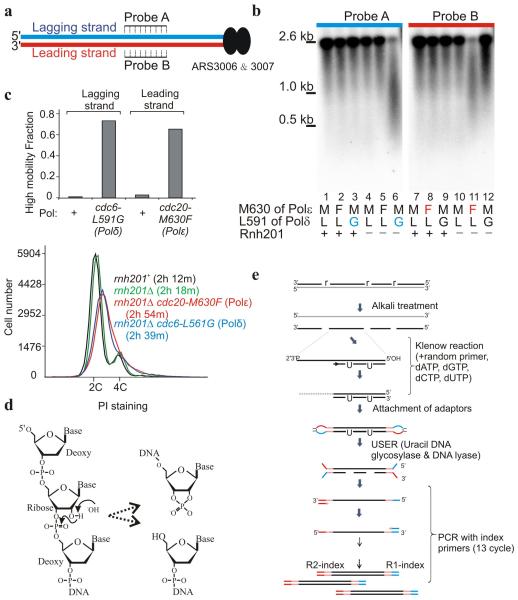
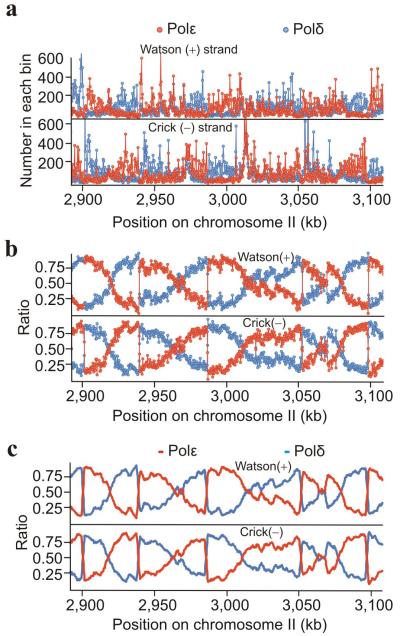
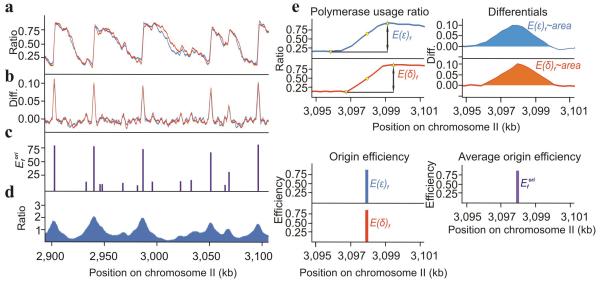
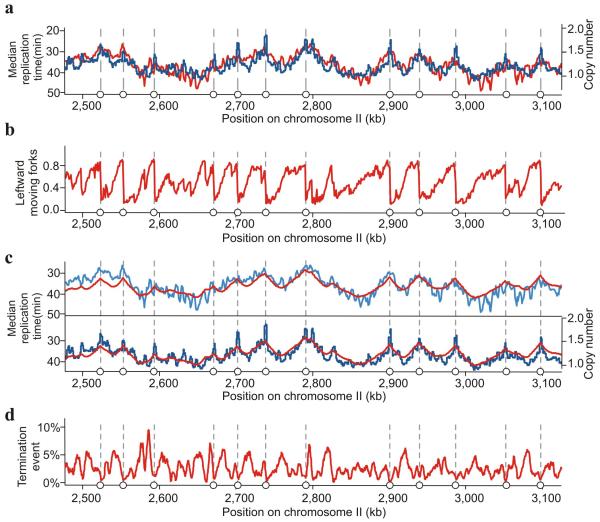
Three eukaryotic DNA polymerases are essential for genome replication. Polymerase (Pol) α-primase initiates each synthesis event and is rapidly replaced by processive DNA polymerases: Polɛ replicates the leading strand, whereas Polδ performs lagging-strand synthesis. However, it is not known whether this division of labor is maintained across the whole genome or how uniform it is within single replicons. Using Schizosaccharomyces pombe, we have developed a polymerase usage sequencing (Pu-seq) strategy to map polymerase usage genome wide. Pu-seq provides direct replication-origin location and efficiency data and indirect estimates of replication timing. We confirm that the division of labor is broadly maintained across an entire genome. However, our data suggest a subtle variability in the usage of the two polymerases within individual replicons. We propose that this results from occasional leading-strand initiation by Polδ followed by exchange for Polɛ.
Figures


References
-
- Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. - PubMed
-
- Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. - PubMed
-
- Stillman B. Origin recognition and the chromosome cycle. FEBS Lett. 2005;579:877–84. - PubMed
References for Online Methods
-
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. - PubMed
-
- Watson AT, Garcia V, Bone N, Carr AM, Armstrong J. Gene tagging and gene replacement using recombinase-mediated cassette exchange in Schizosaccharomyces pombe. Gene. 2008;407:63–74. - PubMed
-
- Lipkin D, Talbert PT, Cohn M. The Mechanism of the Alkaline Hydrolysis of Ribonucleic Acids. J. Am. Chem. Soc. 1954;76:2871–2872.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
