

Sickle cell disease: renal manifestations and mechanisms
- PMID: 25668001
- PMCID: PMC4701210
- DOI: 10.1038/nrneph.2015.8
Sickle cell disease: renal manifestations and mechanisms
Abstract
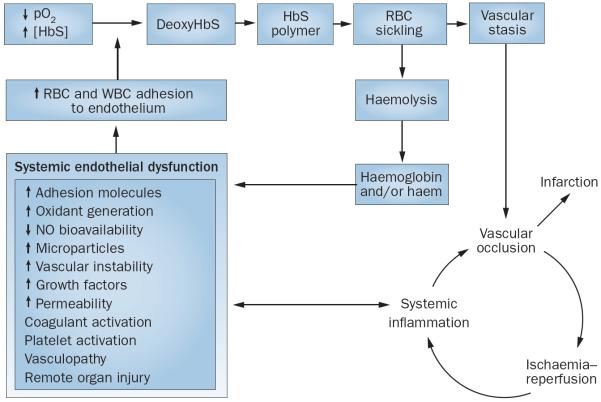
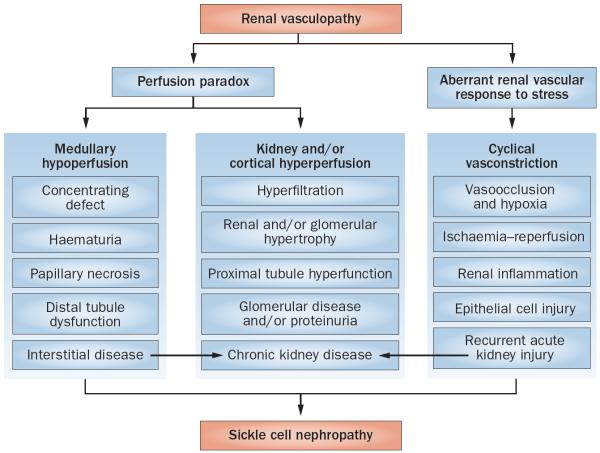
Sickle cell disease (SCD) substantially alters renal structure and function, and causes various renal syndromes and diseases. Such diverse renal outcomes reflect the uniquely complex vascular pathobiology of SCD and the propensity of red blood cells to sickle in the renal medulla because of its hypoxic, acidotic, and hyperosmolar conditions. Renal complications and involvement in sickle cell nephropathy (SCN) include altered haemodynamics, hypertrophy, assorted glomerulopathies, chronic kidney disease, acute kidney injury, impaired urinary concentrating ability, distal nephron dysfunction, haematuria, and increased risks of urinary tract infections and renal medullary carcinoma. SCN largely reflects an underlying vasculopathy characterized by cortical hyperperfusion, medullary hypoperfusion, and an increased, stress-induced vasoconstrictive response. Renal involvement is usually more severe in homozygous disease (sickle cell anaemia, HbSS) than in compound heterozygous types of SCD (for example HbSC and HbSβ(+)-thalassaemia), and is typically mild, albeit prevalent, in the heterozygous state (sickle cell trait, HbAS). Renal involvement contributes substantially to the diminished life expectancy of patients with SCD, accounting for 16-18% of mortality. As improved clinical care promotes survival into adulthood, SCN imposes a growing burden on both individual health and health system costs. This Review addresses the renal manifestations of SCD and focuses on their underlying mechanisms.
Figures
References
-
- Buckalew VM, Jr, Someren A. Renal manifestations of sickle cell disease. Arch. Intern. Med. 1974;133:660–669. - PubMed
-
- Alleyne GA. The kidney in sickle cell anemia. Kidney Int. 1975;7:371–379. - PubMed
-
- de Jong PE, Statius van Eps LW. Sickle cell nephropathy: new insights into its pathophysiology. Kidney Int. 1985;27:711–717. - PubMed
-
- Allon M. Renal abnormalities in sickle cell disease. Arch. Intern. Med. 1990;150:501–504. - PubMed
-
- Pham PT, Pham PC, Wilkinson AH, Lew SQ. Renal abnormalities in sickle cell disease. Kidney Int. 2000;57:1–8. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
