

Environmental genes and genomes: understanding the differences and challenges in the approaches and software for their analyses
- PMID: 25673291
- PMCID: PMC4570204
- DOI: 10.1093/bib/bbv001
Environmental genes and genomes: understanding the differences and challenges in the approaches and software for their analyses
Abstract
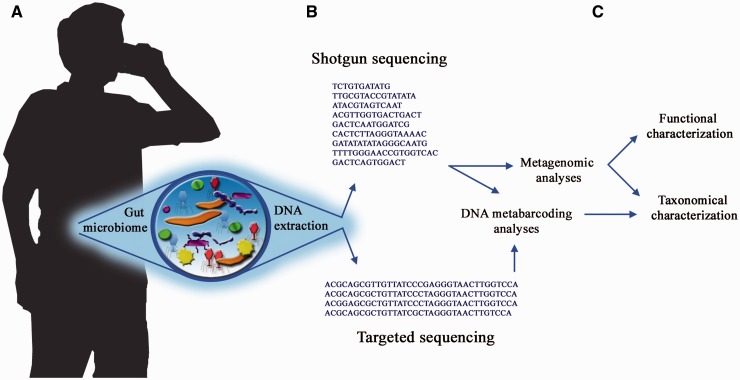
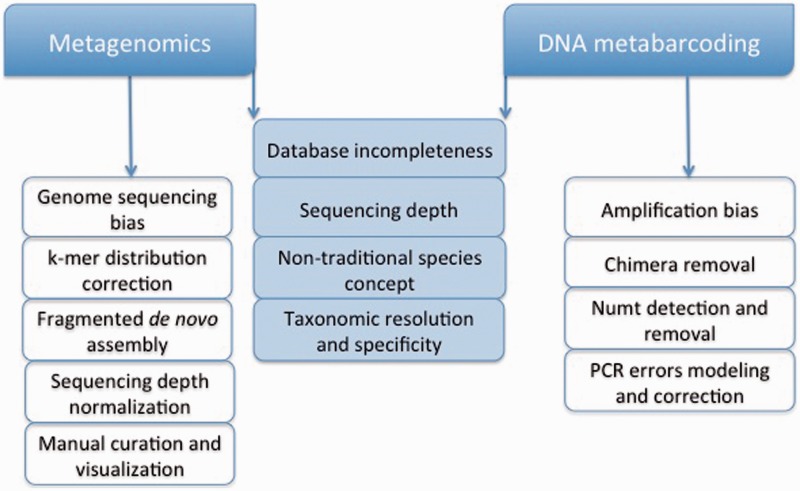
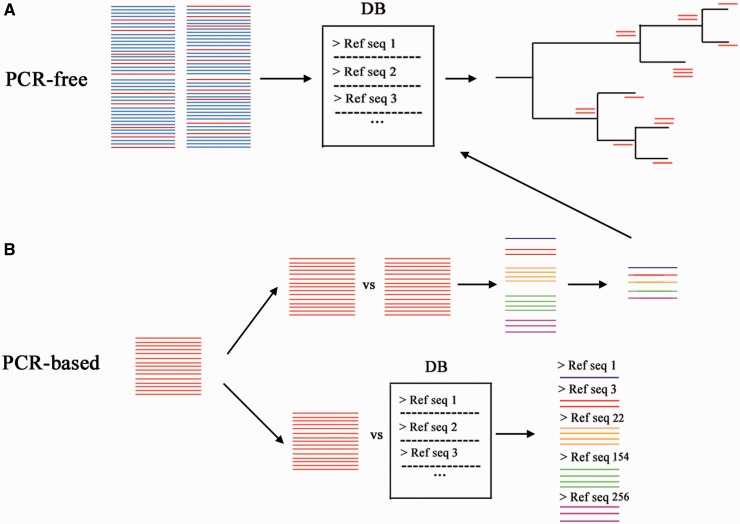
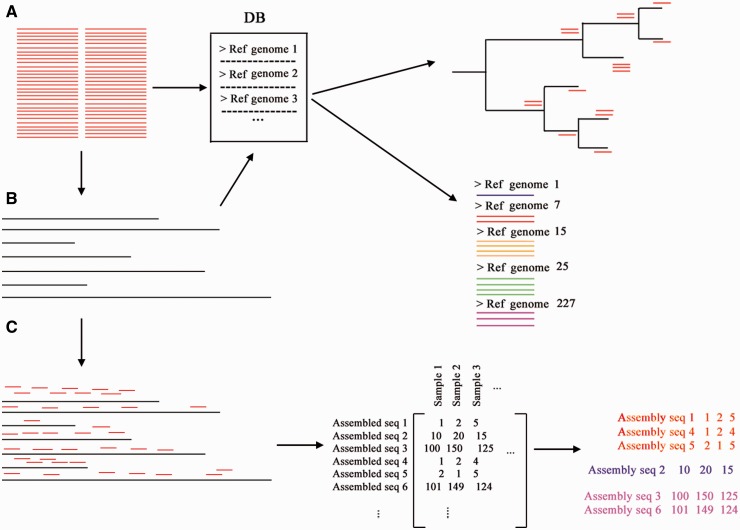
DNA-based taxonomic and functional profiling is widely used for the characterization of organismal communities across a rapidly increasing array of research areas that include the role of microbiomes in health and disease, biomonitoring, and estimation of both microbial and metazoan species richness. Two principal approaches are currently used to assign taxonomy to DNA sequences: DNA metabarcoding and metagenomics. When initially developed, each of these approaches mandated their own particular methods for data analysis; however, with the development of high-throughput sequencing (HTS) techniques they have begun to share many aspects in data set generation and processing. In this review we aim to define the current characteristics, goals and boundaries of each field, and describe the different software used for their analysis. We argue that an appreciation of the potential and limitations of each method can help underscore the improvements required by each field so as to better exploit the richness of current HTS-based data sets.
Keywords: DNA metabarcoding; environment; genome; metagenomics; software development.
© The Author 2015. Published by Oxford University Press.
Figures
References
-
- Eme L, Reigstad LJ, Spang A, et al. Metagenomics of Kamchatkan hot spring filaments reveal two new major (hyper)thermophilic lineages related to Thaumarchaeota. Res Microbiol 2013;164(5):425–38. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
