

Genetic variation analyses of porcine epidemic diarrhea virus isolated in mid-eastern China from 2011 to 2013
- PMID: 25673903
- PMCID: PMC4283240
Genetic variation analyses of porcine epidemic diarrhea virus isolated in mid-eastern China from 2011 to 2013
Abstract
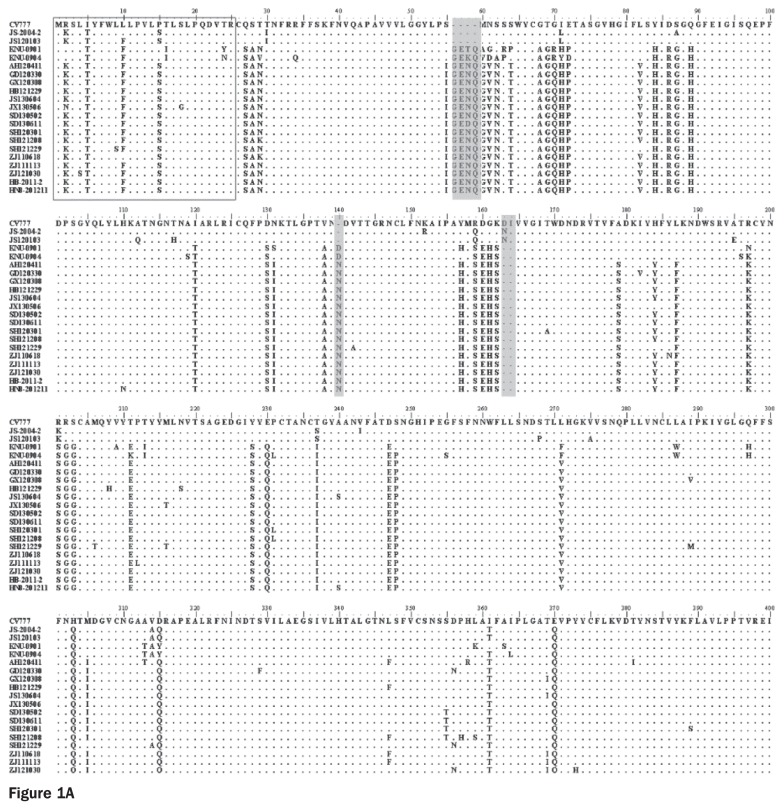
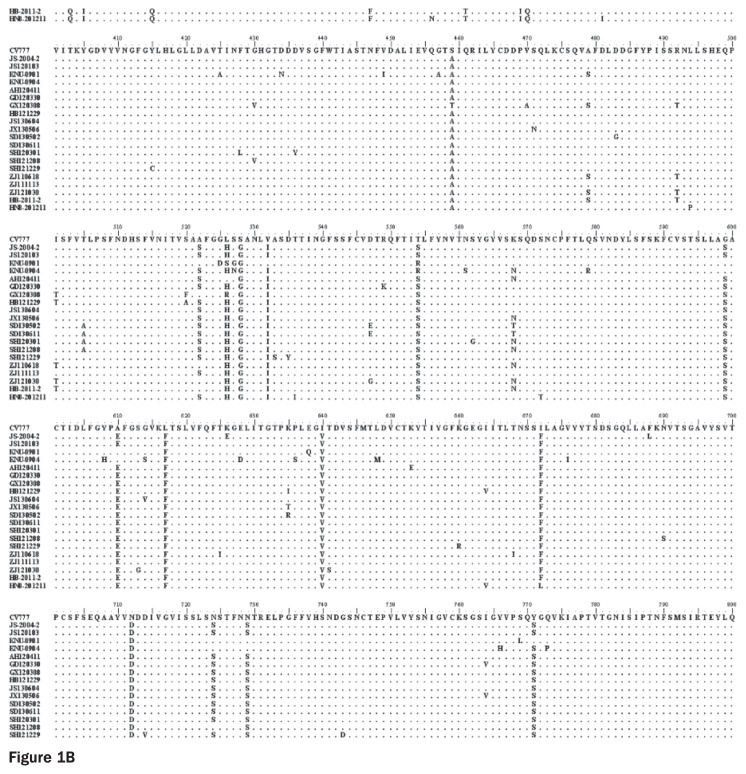
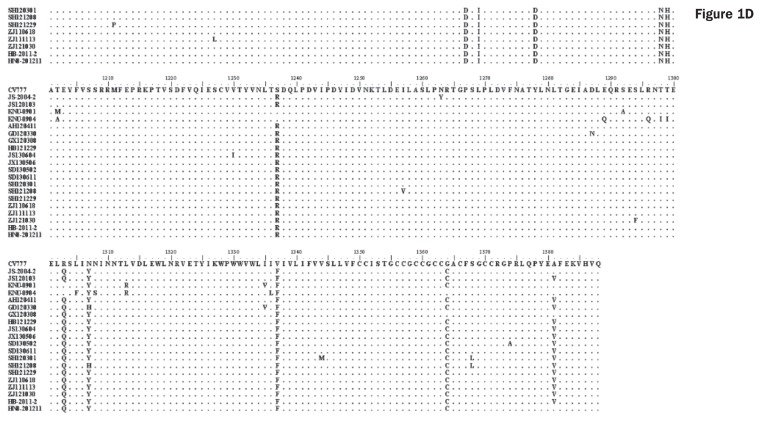
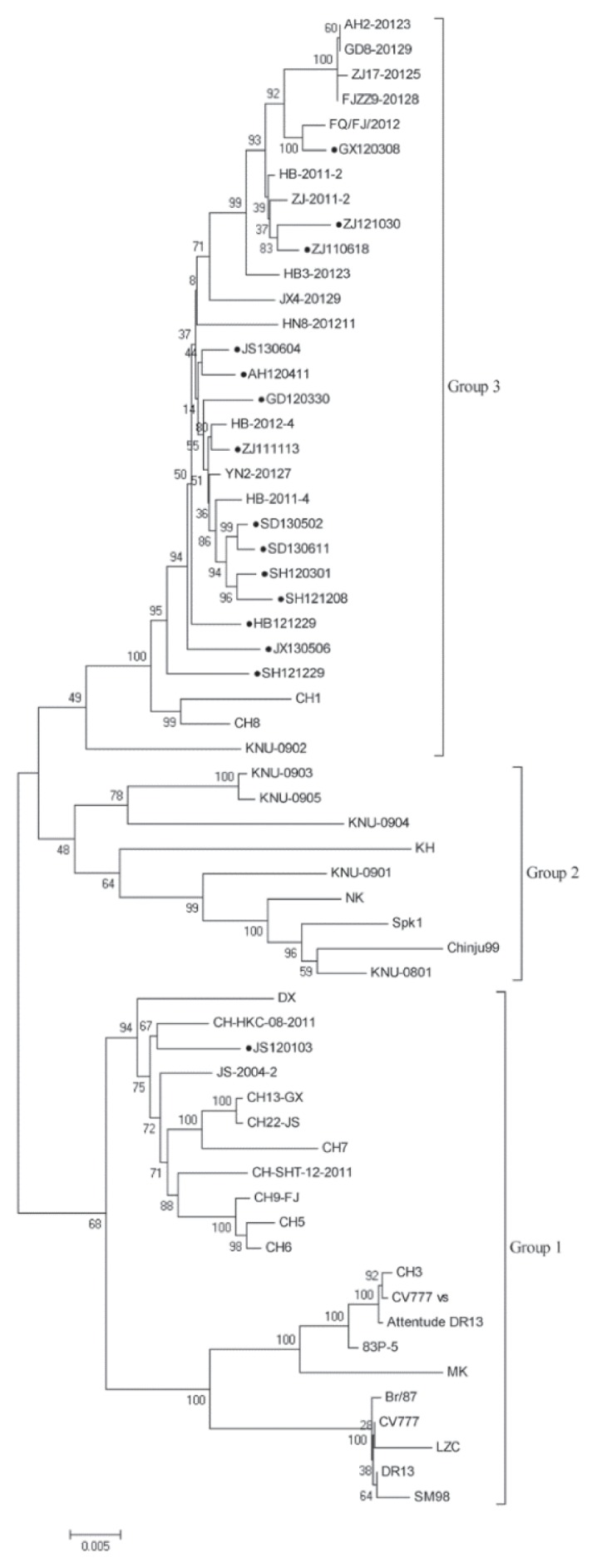
Porcine diarrhea outbreaks caused by porcine epidemic diarrhea virus (PEDV) has occurred in China with significant losses of piglets since 2010. In this study, the complete S and ORF3 genes of 15 field PEDV isolates in mid-eastern China from 2011 to 2013 were detected and compared with other reference strains. Based on S gene, all of the PEDV strains could be assigned to 3 genogroups. Only 1 isolate, JS120103, belonged to genogroup 1 and showed a close relationship with previous Chinese strains DX and JS-2004-2, European strain CV777, and Korean strain DR13. The other 14 isolates belonged to genogroup 3 and showed a close relationship with other Chinese strains isolated after 2010. The S genes of those isolates were 9 nucleotides longer in length than JS120103 and the other reference strains in genogroup 1, with 15 bp insertion and 6 bp deletion. Homology analyses revealed that all of the Chinese field isolates, except JS120103, are 97.6% to 100% (95.8% to 100%) identical in nucleotide (deduced amino acid) sequence to each other. Meanwhile, based on the ORF3 gene, all of the PEDV isolates could be separated into 3 genogroups. Eleven of the 15 field isolates in this study belonged to genogroup 3 and were 95.8% to 100% identical in nucleotide sequence or 95.6% to 100% in deduced amino acid sequence to each other. Our results indicate that the variant PEDV strain spread wildly in mid-eastern China. This will be useful to take into consideration in the control and prevention of this disease.
Des épidémies de diarrhée porcine due au virus de la diarrhée épidémique porcine (VDEP) sont présentes en Chine et ont causé des pertes significatives de porcelets depuis 2010. Dans la présente étude, les gènes S et ORF3 de 15 isolats de champs de VDEP provenant de la portion moyen-orientale de la Chine isolés entre 2011 et 2013 furent détectés et comparés à des souches de référence. Sur la base du gène S, toutes les souches de VDEP pouvaient être réparties dans trois génogroupes. Un seul isolat, JS120103, appartenait au génogroupe 1 et montrait une proche parenté avec les souches chinoises précédentes DX et JS-2004-2, la souche européenne CV777, et la souche coréenne DR13. Les 14 autres isolats appartenaient au génogroupe 3, et montraient une proche parenté avec d’autres souches chinoises isolées après 2010. Les gènes S de ces isolats étaient plus longs de 9 nucléotides que JS120103 et les autres souches de référence du génogroupe 1, avec une insertion de 15 paires de base et une délétion de 6 paires de base. Les analyses d’homologie ont révélé que tous les isolats de champs chinois, sauf JS120103, sont 97,6 % à 100 % (95,8 % à 100 %) identiques entre eux en séquence de nucléotides (d’acides aminés déduits). Également, sur la base du gène ORF3, tous les isolats de VDEP pouvaient être séparés en 3 génogroupes. Onze des 15 isolats de champs de la présente étude appartenaient au génogroupe 3 et étaient 95,8 % à 100 % identiques entre eux en séquence de nucléotides ou 95,6 % à 100 % en séquence d’acides aminés déduits. Ces résultats indiquent que les variants des souches de VDEP sont largement dispersés dans la partie moyenne-orientale de la Chine. Ces données seront utiles à prendre en considération pour la prévention et la maitrise de cette maladie.(Traduit par Docteur Serge Messier).
Figures


Similar articles
-
Detection and phylogenetic analysis of porcine epidemic diarrhea virus in central China based on the ORF3 gene and the S1 gene.Virol J. 2016 Nov 25;13(1):192. doi: 10.1186/s12985-016-0646-8. Virol J. 2016. PMID: 27887624 Free PMC article.
-
Detection and phylogenetic analyses of spike genes in porcine epidemic diarrhea virus strains circulating in China in 2016-2017.Virol J. 2017 Oct 10;14(1):194. doi: 10.1186/s12985-017-0860-z. Virol J. 2017. PMID: 29017599 Free PMC article.
-
Molecular characteristics of the spike gene of porcine epidemic diarrhoea virus strains in Eastern China in 2016.Virus Res. 2018 Mar 2;247:47-54. doi: 10.1016/j.virusres.2018.01.013. Epub 2018 Feb 3. Virus Res. 2018. PMID: 29412159
-
Epidemiology of porcine epidemic diarrhea virus among Chinese pig populations: A meta-analysis.Microb Pathog. 2019 Apr;129:43-49. doi: 10.1016/j.micpath.2019.01.017. Epub 2019 Jan 23. Microb Pathog. 2019. PMID: 30682525
-
Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus.Virol J. 2015 Dec 22;12:193. doi: 10.1186/s12985-015-0421-2. Virol J. 2015. PMID: 26689811 Free PMC article. Review.
Cited by
-
Genomic Motifs as a Novel Indicator of the Relationship between Strains Isolated from the Epidemic of Porcine Epidemic Diarrhea in 2013-2014.PLoS One. 2016 Jan 25;11(1):e0147994. doi: 10.1371/journal.pone.0147994. eCollection 2016. PLoS One. 2016. PMID: 26808527 Free PMC article.
-
Detection and Phylogenetic Analysis of an Exotic Strain of Porcine Epidemic Diarrhea Virus and Its Effect on an Affected Herd Immunized Against the Endemic Strain in Thailand.Animals (Basel). 2025 Jan 15;15(2):225. doi: 10.3390/ani15020225. Animals (Basel). 2025. PMID: 39858225 Free PMC article.
-
Novel Approach for Isolation and Identification of Porcine Epidemic Diarrhea Virus (PEDV) Strain NJ Using Porcine Intestinal Epithelial Cells.Viruses. 2017 Jan 21;9(1):19. doi: 10.3390/v9010019. Viruses. 2017. PMID: 28117718 Free PMC article.
-
Trypsin-independent porcine epidemic diarrhea virus US strain with altered virus entry mechanism.BMC Vet Res. 2017 Nov 25;13(1):356. doi: 10.1186/s12917-017-1283-1. BMC Vet Res. 2017. PMID: 29178878 Free PMC article.
-
Molecular characterization of US-like and Asian non-S INDEL strains of porcine epidemic diarrhea virus (PEDV) that circulated in Japan during 2013-2016 and PEDVs collected from recurrent outbreaks.BMC Vet Res. 2018 Mar 14;14(1):96. doi: 10.1186/s12917-018-1409-0. BMC Vet Res. 2018. PMID: 29540176 Free PMC article.
References
-
- Bridgen A, Kocherhans R, Tobler K, Carvajal A, Ackermann M. Further analysis of the genome of porcine epidemic diarrhoea virus. Adv Exp Med Biol. 1998;440:781–786. - PubMed
-
- Chang SH, Bae JL, Kang TJ, et al. Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells. 2002;14:295–299. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources