

Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles
- PMID: 25675496
- PMCID: PMC4345584
- DOI: 10.1073/pnas.1417683112
Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles
Abstract
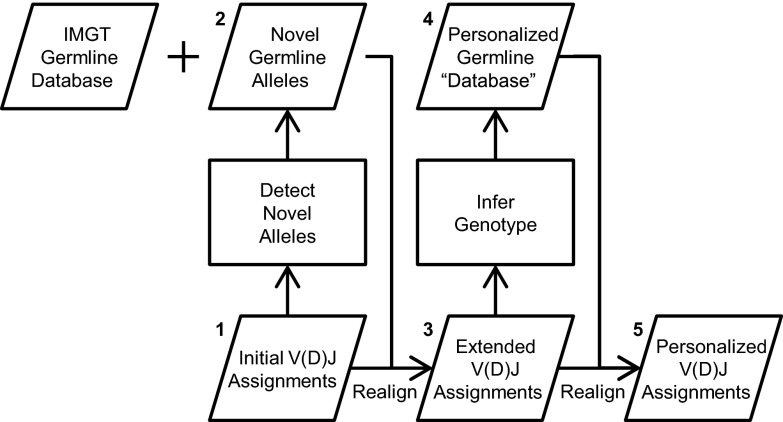
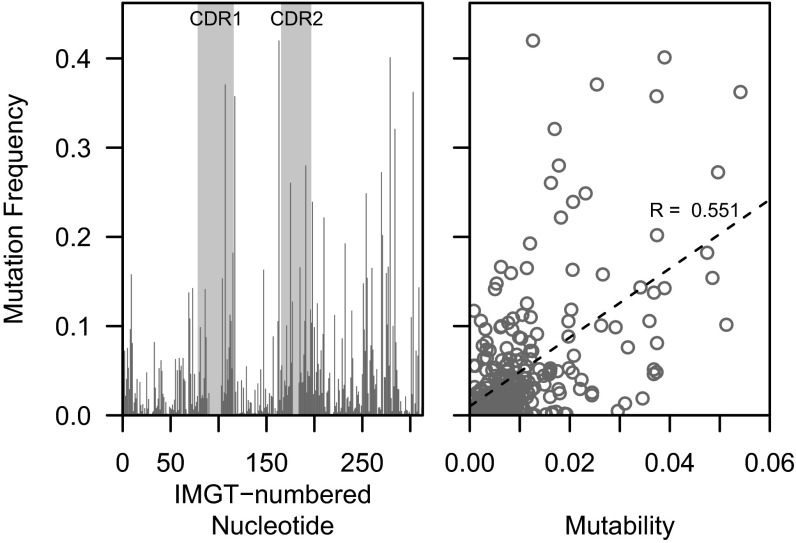
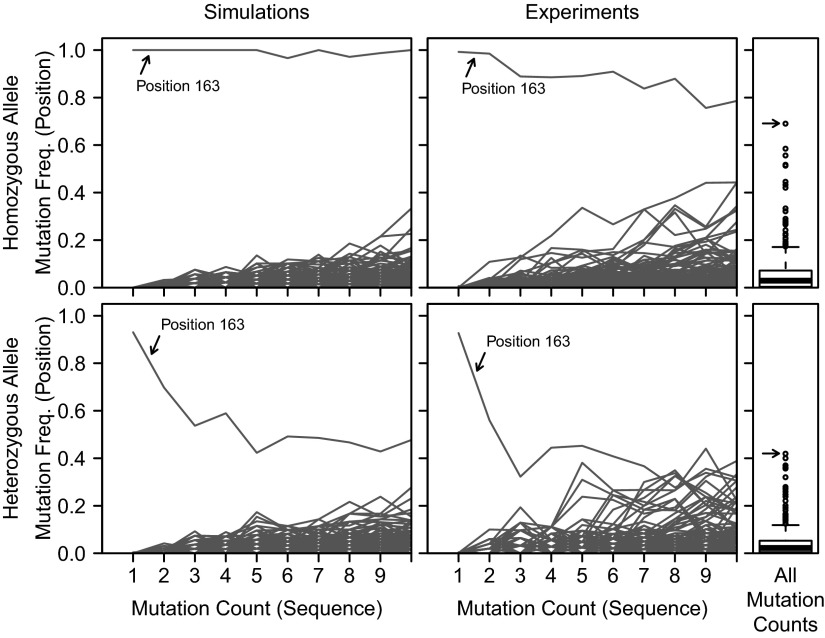
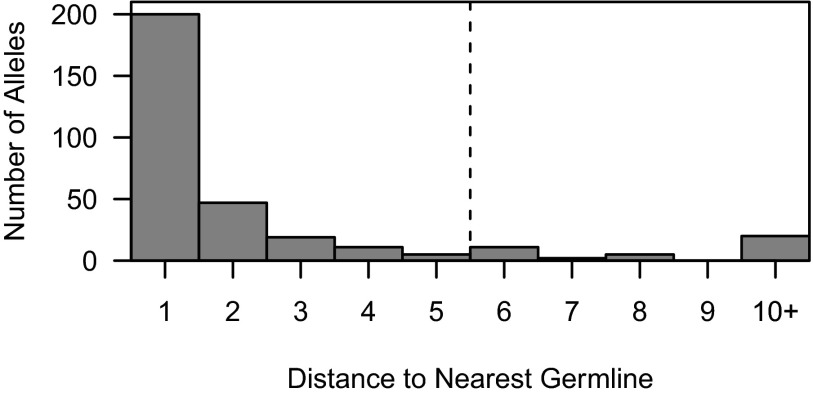
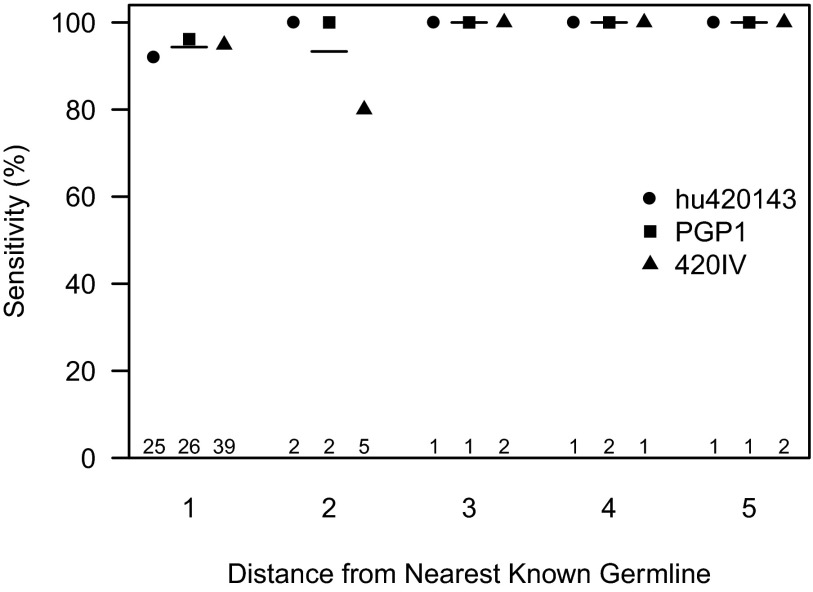
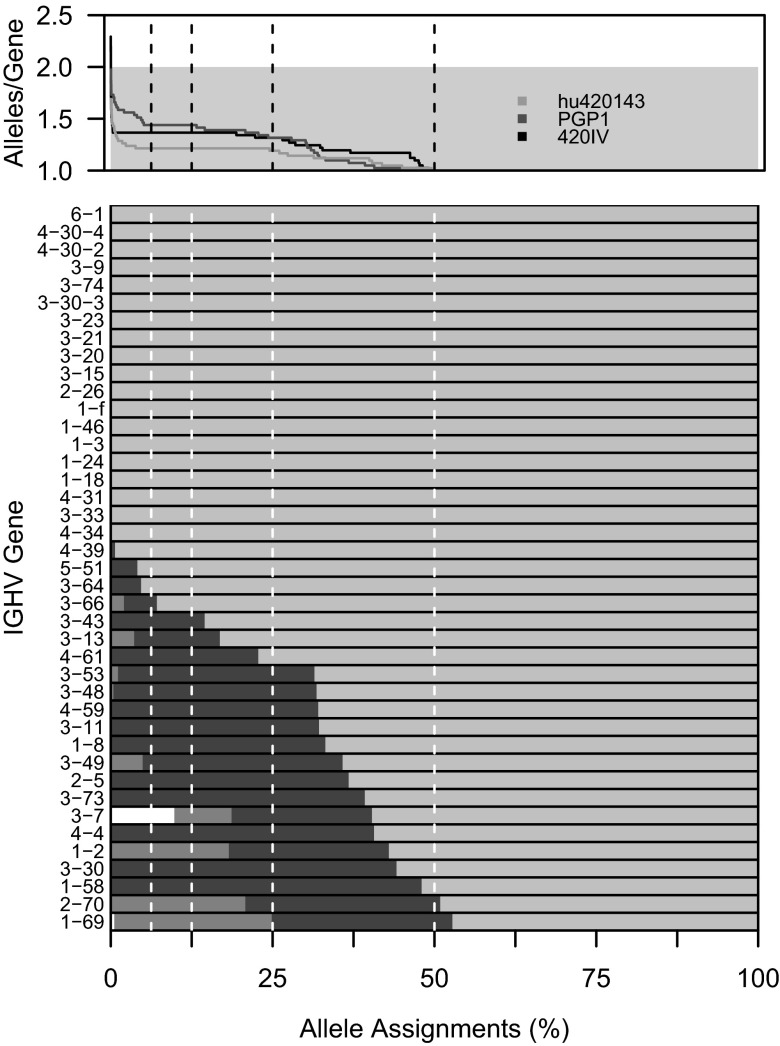
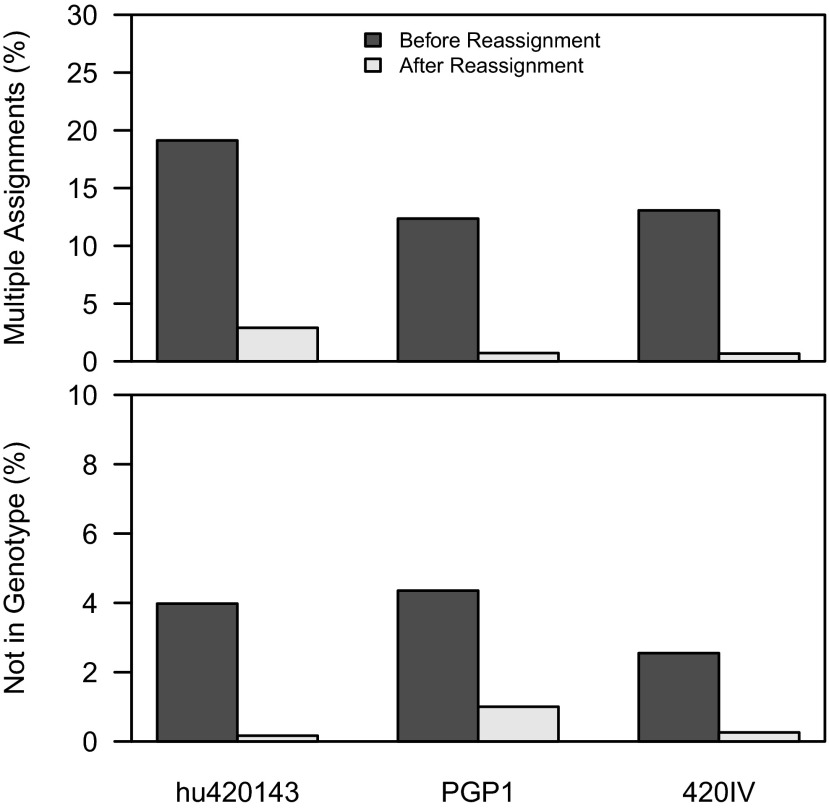
Individual variation in germline and expressed B-cell immunoglobulin (Ig) repertoires has been associated with aging, disease susceptibility, and differential response to infection and vaccination. Repertoire properties can now be studied at large-scale through next-generation sequencing of rearranged Ig genes. Accurate analysis of these repertoire-sequencing (Rep-Seq) data requires identifying the germline variable (V), diversity (D), and joining (J) gene segments used by each Ig sequence. Current V(D)J assignment methods work by aligning sequences to a database of known germline V(D)J segment alleles. However, existing databases are likely to be incomplete and novel polymorphisms are hard to differentiate from the frequent occurrence of somatic hypermutations in Ig sequences. Here we develop a Tool for Ig Genotype Elucidation via Rep-Seq (TIgGER). TIgGER analyzes mutation patterns in Rep-Seq data to identify novel V segment alleles, and also constructs a personalized germline database containing the specific set of alleles carried by a subject. This information is then used to improve the initial V segment assignments from existing tools, like IMGT/HighV-QUEST. The application of TIgGER to Rep-Seq data from seven subjects identified 11 novel V segment alleles, including at least one in every subject examined. These novel alleles constituted 13% of the total number of unique alleles in these subjects, and impacted 3% of V(D)J segment assignments. These results reinforce the highly polymorphic nature of human Ig V genes, and suggest that many novel alleles remain to be discovered. The integration of TIgGER into Rep-Seq processing pipelines will increase the accuracy of V segment assignments, thus improving B-cell repertoire analyses.
Keywords: B-cell repertoire; adaptive immunity; next-generation sequencing; somatic hypermutation; variable gene segment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lefranc MP. Nomenclature of the human immunoglobulin heavy (IGH) genes. Exp Clin Immunogenet. 2001;18(2):100–116. - PubMed
-
- Muramatsu M, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. - PubMed
-
- Papavasiliou FN, Schatz DG. Somatic hypermutation of immunoglobulin genes: Merging mechanisms for genetic diversity. Cell. 2002;109(Suppl):S35–S44. - PubMed
-
- Watson CT, Breden F. The immunoglobulin heavy chain locus: Genetic variation, missing data, and implications for human disease. Genes Immun. 2012;13(5):363–373. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
