

Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2
- PMID: 25679999
- PMCID: PMC4332675
- DOI: 10.1371/journal.pone.0117158
Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2
Erratum in
-
Correction: Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2.PLoS One. 2015 May 7;10(5):e0125310. doi: 10.1371/journal.pone.0125310. eCollection 2015. PLoS One. 2015. PMID: 25951182 Free PMC article. No abstract available.
Abstract
Distal arthrogryposes (DAs) are a group of disorders that mainly involve the distal parts of the limbs and at least ten different DAs have been described to date. DAs are mostly described as autosomal dominant disorders with variable expressivity and incomplete penetrance, but recently autosomal recessive pattern was reported in distal arthrogryposis type 5D. Mutations in the contractile genes are found in about 50% of all DA patients. Of these genes, mutations in the gene encoding myosin binding protein C slow MYBPC1 were recently identified in two families with distal arthrogryposis type 1B. Here, we described two large Chinese families with autosomal dominant distal arthrogryposis type 2(DA2) with incomplete penetrance and variable expressivity. Some unique overextension contractures of the lower limbs and some distinctive facial features were present in our DA2 pedigrees. We performed follow-up DNA sequencing after linkage mapping and first identified two novel MYBPC1 mutations (c.1075G>A [p.E359K] and c.956C>T [p.P319L]) responsible for these Chinese DA2 families of which one introduced by germline mosacism. Each mutation was found to cosegregate with the DA2 phenotype in each family but not in population controls. Both substitutions occur within C2 immunoglobulin domain, which together with C1 and the M motif constitute the binding site for the S2 subfragment of myosin. Our results expand the phenotypic spectrum of MYBPC1-related arthrogryposis multiplex congenita (AMC). We also proposed the possible molecular mechanisms that may underlie the pathogenesis of DA2 myopathy associated with these two substitutions in MYBPC1.
Conflict of interest statement
Figures
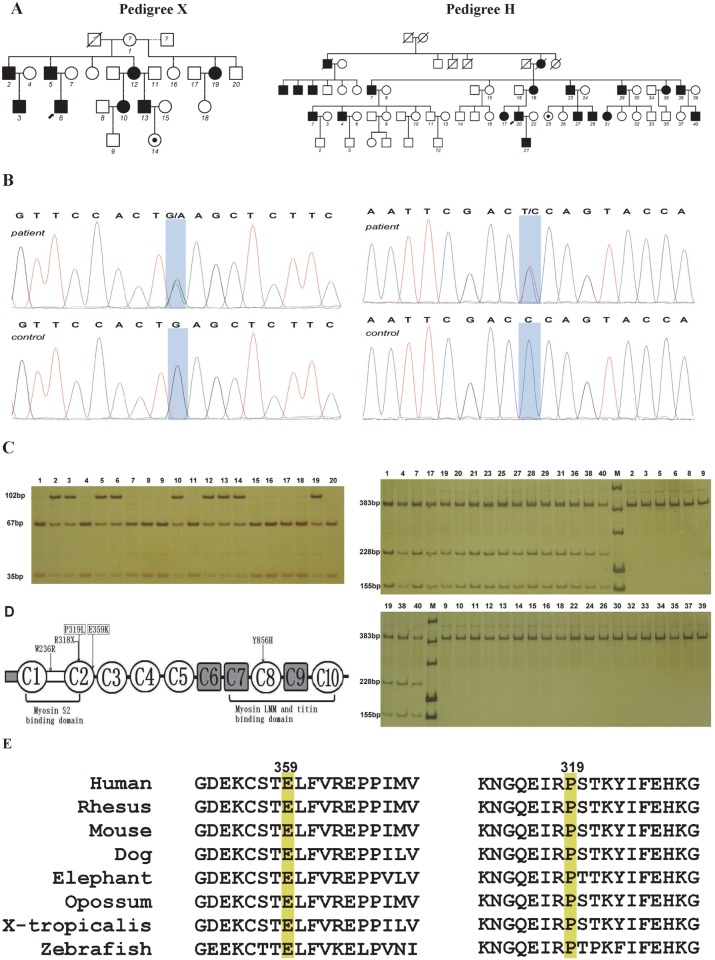
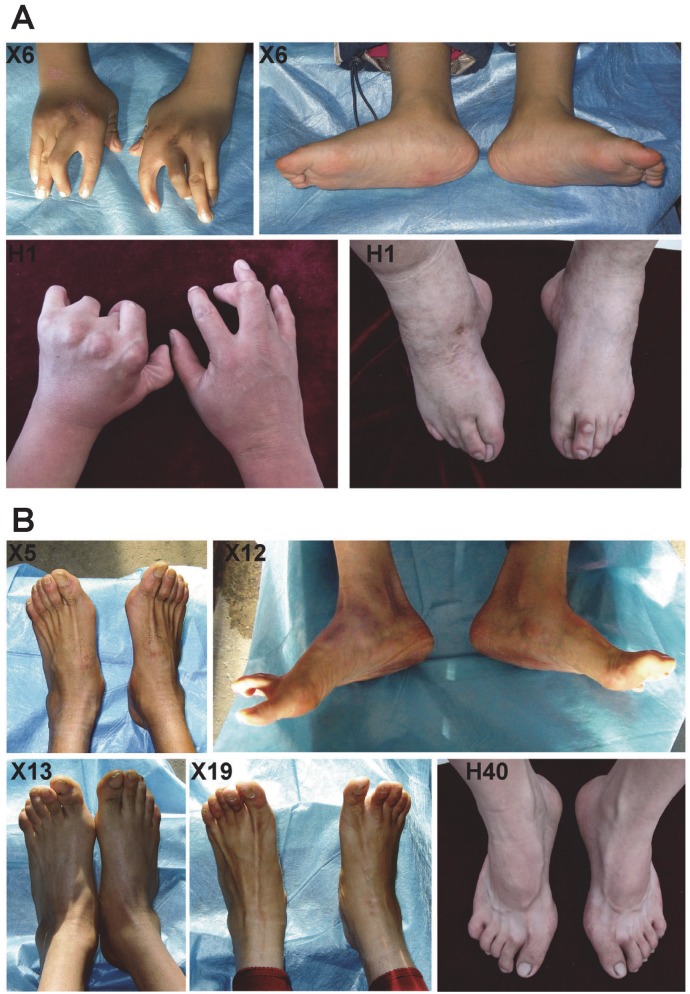
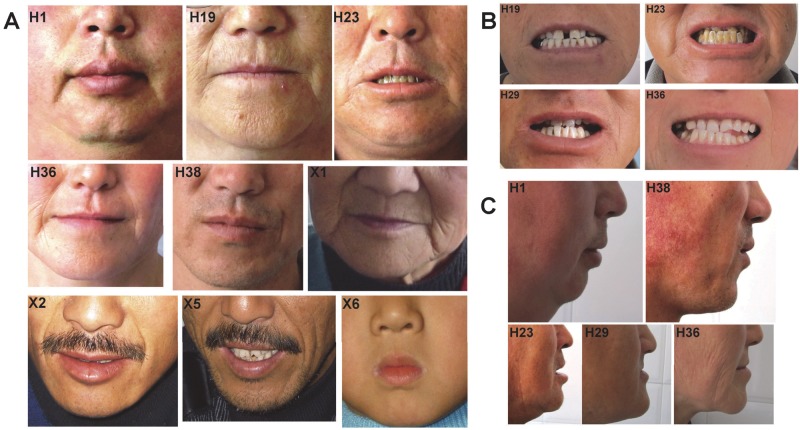
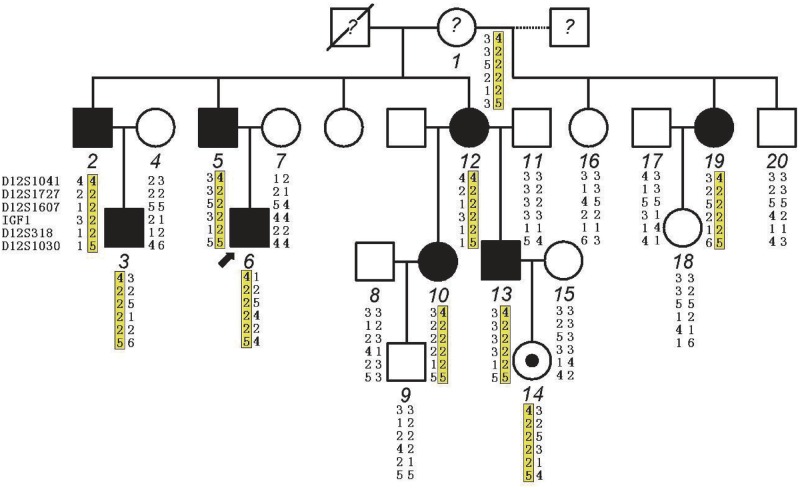
References
-
- Bamshad M, Jorde LB, Carey JC (1996) A revised and extended classification of the distal arthrogryposes. Am J Med Genet 65: 277–281. - PubMed
-
- Hall JG, Reed SD, Greene G (1982) The distal arthrogryposes: delineation of new entities—review and nosologic discussion. Am J Med Genet 11: 185–239. - PubMed
-
- Stevenson DA, Carey JC, Palumbos J, Rutherford A, Dolcourt J, et al. (2006) Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics 117: 754–762. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
