

Analysis of HGD Gene Mutations in Patients with Alkaptonuria from the United Kingdom: Identification of Novel Mutations
- PMID: 25681086
- PMCID: PMC4582018
- DOI: 10.1007/8904_2014_380
Analysis of HGD Gene Mutations in Patients with Alkaptonuria from the United Kingdom: Identification of Novel Mutations
Abstract
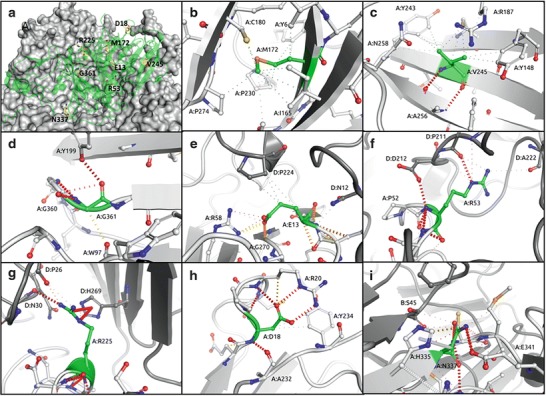
Alkaptonuria (AKU) is a rare autosomal recessive disorder with incidence ranging from 1:100,000 to 1:250,000. The disorder is caused by a deficiency of the enzyme homogentisate 1,2-dioxygenase (HGD), which results from defects in the HGD gene. This enzyme converts homogentisic acid to maleylacetoacetate and has a major role in the catabolism of phenylalanine and tyrosine. To elucidate the mutation spectrum of the HGD gene in patients with alkaptonuria from 42 patients attending the National Alkaptonuria Centre, 14 exons of the HGD gene and the intron-exon boundaries were analysed by PCR-based sequencing. A total of 34 sequence variants was observed, confirming the genetic heterogeneity of AKU. Of these mutations, 26 were missense substitutions and four splice site mutations. There were two deletions and one duplication giving rise to frame shifts and one substitution abolishing the translation termination codon (no stop). Nine of the mutations were previously unreported novel variants. Using computational approaches based on the 3D structure, these novel mutations are predicted to affect the activity of the protein complex through destabilisation of the individual protomer structure or through disruption of protomer-protomer interactions.
Keywords: Alkaptonuria; Homogentisic acid; Novel mutation; Rare genetic disorder; Sequencing.
Figures
References
-
- Davison AS, Milan AM, Hughes AT, Dutton JJ, Ranganath LR (2014) Serum concentrations and urinary excretion of homogentisic acid and tyrosine in normal subjects. Clin Chem Lab Med. doi:10.1515/cclm-2014-0668 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
