

Pathobiology of liver fibrosis: a translational success story
- PMID: 25681399
- PMCID: PMC4477794
- DOI: 10.1136/gutjnl-2014-306842
Pathobiology of liver fibrosis: a translational success story
Erratum in
-
Correction.Gut. 2015 Aug;64(8):1337. doi: 10.1136/gutjnl-2014-306842corr1. Gut. 2015. PMID: 26160826 No abstract available.
Abstract
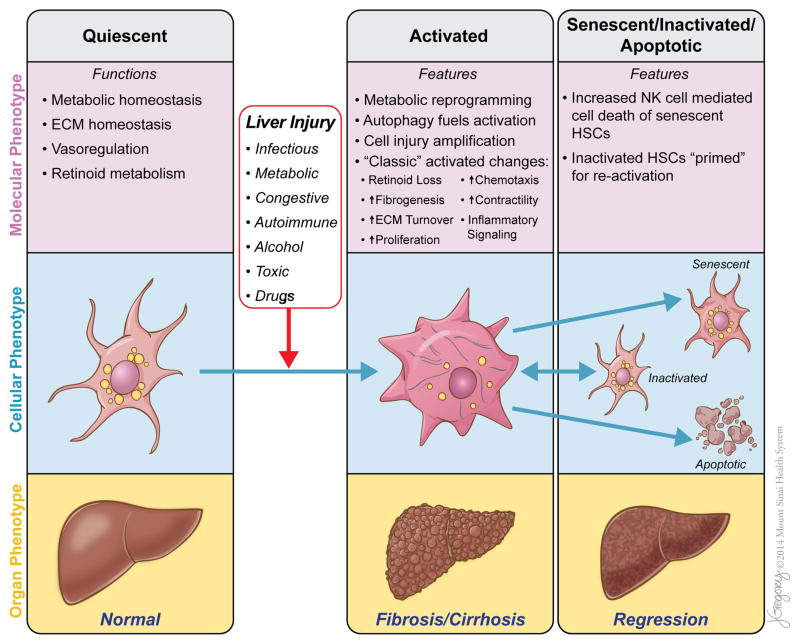
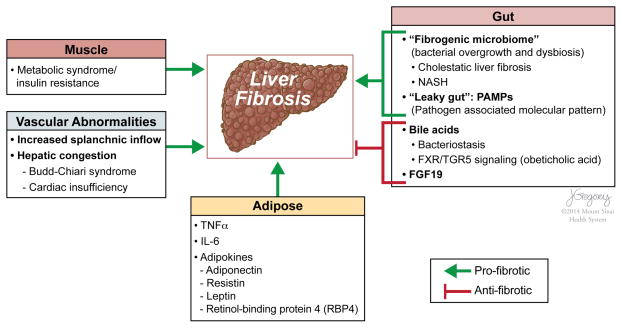
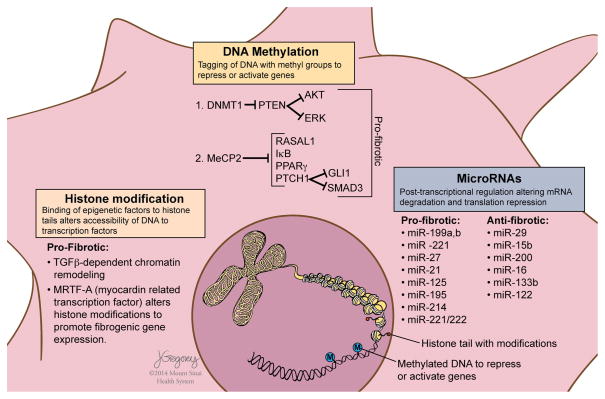
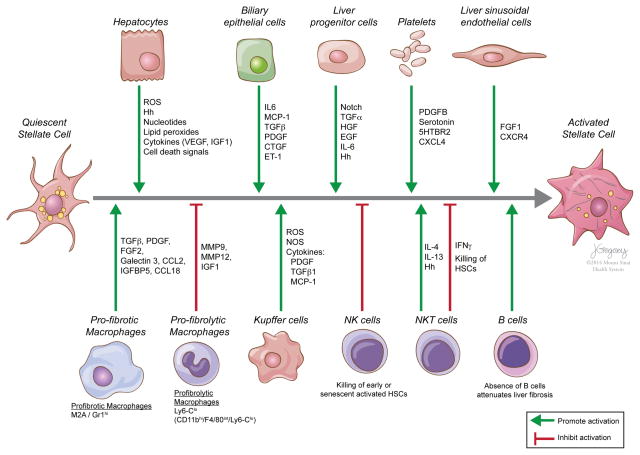
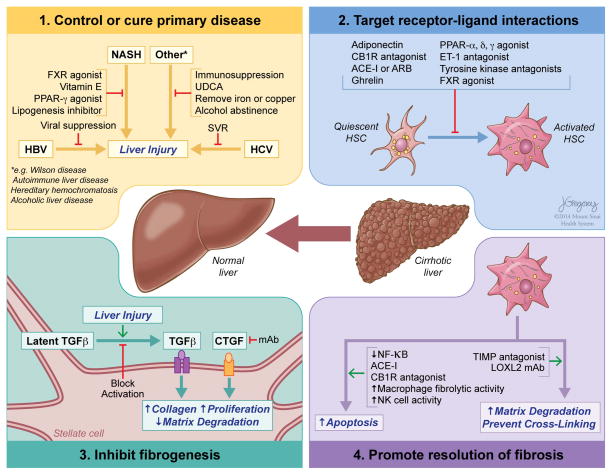
Reversibility of hepatic fibrosis and cirrhosis following antiviral therapy for hepatitis B or C has advanced the prospect of developing antifibrotic therapies for patients with chronic liver diseases, especially non-alcoholic steatohepatitis. Mechanisms of fibrosis have focused on hepatic stellate cells, which become fibrogenic myofibroblasts during injury through 'activation', and are at the nexus of efforts to define novel drug targets. Recent studies have clarified pathways of stellate cell gene regulation and epigenetics, emerging pathways of fibrosis regression through the recruitment and amplification of fibrolytic macrophages, nuanced responses of discrete inflammatory cell subsets and the identification of the 'ductular reaction' as a marker of severe injury and repair. Based on our expanded knowledge of fibrosis pathogenesis, attention is now directed towards strategies for antifibrotic therapies and regulatory challenges for conducting clinical trials with these agents. New therapies are attempting to: 1) Control or cure the primary disease or reduce tissue injury; 2) Target receptor-ligand interactions and intracellular signaling; 3) Inhibit fibrogenesis; and 4) Promote resolution of fibrosis. Progress is urgently needed in validating non-invasive markers of fibrosis progression and regression that can supplant biopsy and shorten the duration of clinical trials. Both scientific and clinical challenges remain, however the past three decades of steady progress in understanding liver fibrosis have contributed to an emerging translational success story, with realistic hopes for antifibrotic therapies to treat patients with chronic liver disease in the near future.
Keywords: CIRRHOSIS; EXTRACELLULAR MATRIX; FATTY LIVER; HEPATIC FIBROSIS; HEPATIC STELLATE CELL.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://group.bmj.com/group/rights-licensing/permissions.
Conflict of interest statement
Figures
Comment in
-
Quantitative liver MRI including extracellular volume fraction for non-invasive quantification of liver fibrosis: a prospective proof-of-concept study.Gut. 2018 Mar;67(3):593-594. doi: 10.1136/gutjnl-2017-314561. Epub 2017 Jul 28. Gut. 2018. PMID: 28754777 No abstract available.
References
-
- Perez-Tamayo R. Cirrhosis of the liver: a reversible disease? Pathol Annu. 1979;14 (Pt 2):183–213. - PubMed
-
- Okazaki I, Maruyama K. Collagenase activity in experimental hepatic fibrosis. Nature. 1974;252:49–50. - PubMed
-
- Pellicoro A, Ramachandran P, Iredale JP, et al. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181–94. - PubMed
-
- Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381:468–75. - PubMed
-
- D’Ambrosio R, Aghemo A, Rumi MG, et al. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology. 2012;56:532–43. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous