

De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay
- PMID: 25683120
- PMCID: PMC4375444
- DOI: 10.1016/j.ajhg.2015.01.003
De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay
Abstract
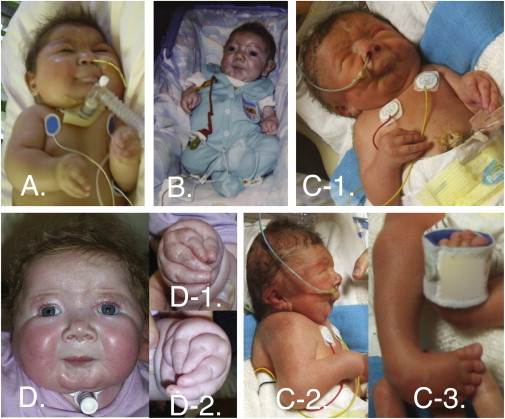
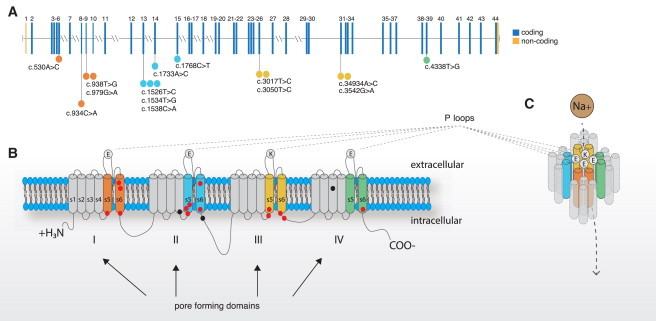
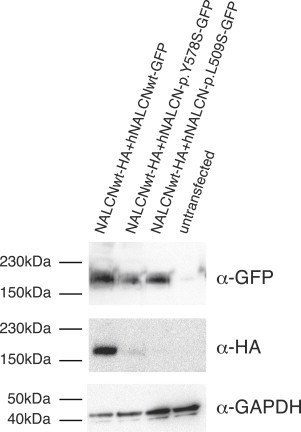
Freeman-Sheldon syndrome, or distal arthrogryposis type 2A (DA2A), is an autosomal-dominant condition caused by mutations in MYH3 and characterized by multiple congenital contractures of the face and limbs and normal cognitive development. We identified a subset of five individuals who had been putatively diagnosed with "DA2A with severe neurological abnormalities" and for whom congenital contractures of the limbs and face, hypotonia, and global developmental delay had resulted in early death in three cases; this is a unique condition that we now refer to as CLIFAHDD syndrome. Exome sequencing identified missense mutations in the sodium leak channel, non-selective (NALCN) in four families affected by CLIFAHDD syndrome. We used molecular-inversion probes to screen for NALCN in a cohort of 202 distal arthrogryposis (DA)-affected individuals as well as concurrent exome sequencing of six other DA-affected individuals, thus revealing NALCN mutations in ten additional families with "atypical" forms of DA. All 14 mutations were missense variants predicted to alter amino acid residues in or near the S5 and S6 pore-forming segments of NALCN, highlighting the functional importance of these segments. In vitro functional studies demonstrated that NALCN alterations nearly abolished the expression of wild-type NALCN, suggesting that alterations that cause CLIFAHDD syndrome have a dominant-negative effect. In contrast, homozygosity for mutations in other regions of NALCN has been reported in three families affected by an autosomal-recessive condition characterized mainly by hypotonia and severe intellectual disability. Accordingly, mutations in NALCN can cause either a recessive or dominant condition characterized by varied though overlapping phenotypic features, perhaps based on the type of mutation and affected protein domain(s).
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Toydemir R.M., Rutherford A., Whitby F.G., Jorde L.B., Carey J.C., Bamshad M.J. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 2006;38:561–565. - PubMed
-
- Beck A.E., McMillin M.J., Gildersleeve H.I., Shively K.M., Tang A., Bamshad M.J. Genotype-phenotype relationships in Freeman-Sheldon syndrome. Am. J. Med. Genet. A. 2014;164A:2808–2813. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- U54 HG006493/HG/NHGRI NIH HHS/United States
- RC2 HG005608/HG/NHGRI NIH HHS/United States
- 1R01HD048895/HD/NICHD NIH HHS/United States
- 1RC2HG005608/HG/NHGRI NIH HHS/United States
- R00 HG004316/HG/NHGRI NIH HHS/United States
- K23 HD057331/HD/NICHD NIH HHS/United States
- R01 HD048895/HD/NICHD NIH HHS/United States
- R25 HG007153/HG/NHGRI NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
- 5R000HG004316/HG/NHGRI NIH HHS/United States
- 1U54HG006493/HG/NHGRI NIH HHS/United States
- 5K23HD057331/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
