

Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta
- PMID: 25683121
- PMCID: PMC4375534
- DOI: 10.1016/j.ajhg.2015.01.002
Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta
Abstract
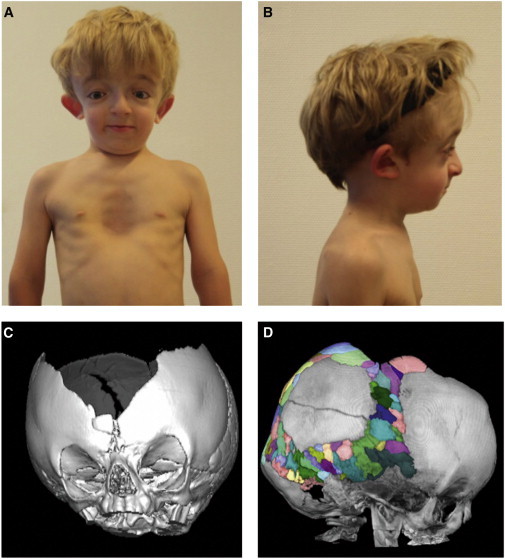
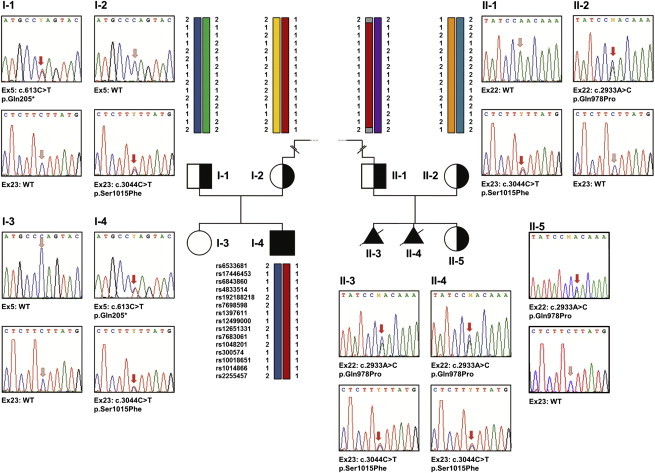
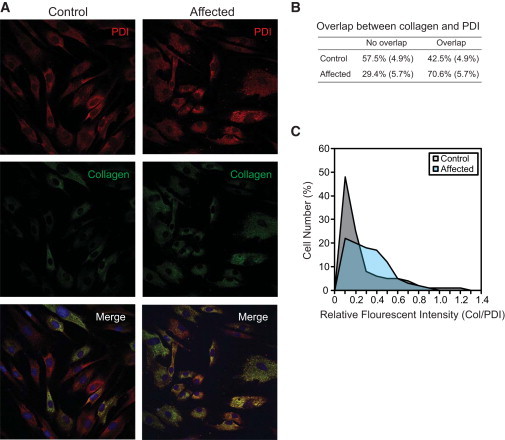
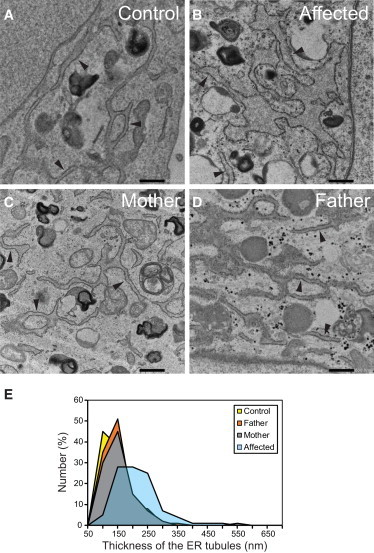
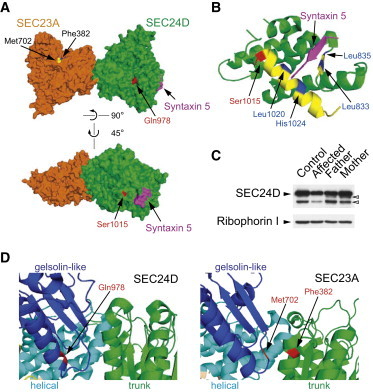
As a result of a whole-exome sequencing study, we report three mutant alleles in SEC24D, a gene encoding a component of the COPII complex involved in protein export from the ER: the truncating mutation c.613C>T (p.Gln205(∗)) and the missense mutations c.3044C>T (p.Ser1015Phe, located in a cargo-binding pocket) and c.2933A>C (p.Gln978Pro, located in the gelsolin-like domain). Three individuals from two families affected by a similar skeletal phenotype were each compound heterozygous for two of these mutant alleles, with c.3044C>T being embedded in a 14 Mb founder haplotype shared by all three. The affected individuals were a 7-year-old boy with a phenotype most closely resembling Cole-Carpenter syndrome and two fetuses initially suspected to have a severe type of osteogenesis imperfecta. All three displayed a severely disturbed ossification of the skull and multiple fractures with prenatal onset. The 7-year-old boy had short stature and craniofacial malformations including macrocephaly, midface hypoplasia, micrognathia, frontal bossing, and down-slanting palpebral fissures. Electron and immunofluorescence microscopy of skin fibroblasts of this individual revealed that ER export of procollagen was inefficient and that ER tubules were dilated, faithfully reproducing the cellular phenotype of individuals with cranio-lentico-sutural dysplasia (CLSD). CLSD is caused by SEC23A mutations and displays a largely overlapping craniofacial phenotype, but it is not characterized by generalized bone fragility and presented with cataracts in the original family described. The cellular and morphological phenotypes we report are in concordance with the phenotypes described for the Sec24d-deficient fish mutants vbi (medaka) and bulldog (zebrafish).
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. - PubMed
-
- Canty E.G., Kadler K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005;118:1341–1353. - PubMed
-
- Byers P.H., Pyott S.M. Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 2012;46:475–497. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- R01 GM110373/GM/NIGMS NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- R21 DE022419/DE/NIDCR NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
